 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a29 | ||||||
|---|---|---|---|---|---|---|---|
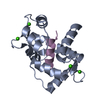
| Title | CALMODULIN COMPLEXED WITH TRIFLUOPERAZINE (1:2 COMPLEX) | ||||||
 Components Components | CALMODULIN | ||||||
 Keywords Keywords | CALCIUM-BINDING PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationcalcineurin-mediated signaling / detection of calcium ion / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / spindle pole / myelin sheath / synaptic vesicle membrane / protein domain specific binding / calcium ion binding / centrosome / protein-containing complex ...calcineurin-mediated signaling / detection of calcium ion / regulation of release of sequestered calcium ion into cytosol by sarcoplasmic reticulum / spindle pole / myelin sheath / synaptic vesicle membrane / protein domain specific binding / calcium ion binding / centrosome / protein-containing complex / cytoplasm / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.74 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.74 Å | ||||||
 Authors Authors | Bocskei, Zs. / Harmat, V. / Vertessy, B.G. / Ovadi, J. / Naray-Szabo, G. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Simultaneous binding of drugs with different chemical structures to Ca2+-calmodulin: crystallographic and spectroscopic studies. Authors: Vertessy, B.G. / Harmat, V. / Bocskei, Z. / Naray-Szabo, G. / Orosz, F. / Ovadi, J. #1: Journal: Biochemistry / Year: 1994Title: Drug Binding by Calmodulin: Crystal Structure of a Calmodulin-Trifluoperazine Complex Authors: Cook, W.J. / Walter, L.J. / Walter, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a29.cif.gz | 40.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a29.ent.gz | 27.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1a29.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a2/1a29ftp://data.pdbj.org/pub/pdb/validation_reports/a2/1a29 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1linS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 16721.350 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) | ||
|---|---|---|---|
| #2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Ca#3: Chemical | 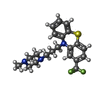  Mass: 407.496 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H24F3N3S Mass: 407.496 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H24F3N3S |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.5 Å3/Da / Density % sol: 52 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5 Details: PROTEIN WAS CRYSTALLIZED BY SITTING DROP TECHNIQUE IN THE COLD ROOM (8 DEGREES C +/- 2 DEGREES C) BY MIXING 4 MICROLITERS OF 1 MM PROTEIN CONTAINING 1.2-1.5 MM TFP IN 5 MM CACL2 WITH 4 ...Details: PROTEIN WAS CRYSTALLIZED BY SITTING DROP TECHNIQUE IN THE COLD ROOM (8 DEGREES C +/- 2 DEGREES C) BY MIXING 4 MICROLITERS OF 1 MM PROTEIN CONTAINING 1.2-1.5 MM TFP IN 5 MM CACL2 WITH 4 MICROLITERS OF THE RESERVOIR SOLUTION (1 ML 10 MM SODIUM CACODYLATE/HCL BUFFER, PH 5.2-5.6 WITH 10 MM CACL2, 25-30 % (W/V) POLYETHYLENE GLYCOL 6000), CRYSTAL GROWTH TOOK 2-3 WEEKS., pH 5.0 Temp details: cold room (8 +/-2 C) | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 6-10 ℃ / pH: 5.2 / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Feb 1, 1995 / Details: NORMAL FOCUS |
| Radiation | Monochromator: GRAPHITE(002) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.74→87.7 Å / Num. obs: 17223 / % possible obs: 98 % / Observed criterion σ(I): 1 / Redundancy: 1.92 % / Biso Wilson estimate: 32 Å2 / Rmerge(I) obs: 0.086 / Rsym value: 0.0737 / Net I/σ(I): 11.3 |
| Reflection shell | Resolution: 2.74→2.86 Å / Redundancy: 1.9 % / Rmerge(I) obs: 0.092 / Mean I/σ(I) obs: 1.84 / Rsym value: 0.205 / % possible all: 98.2 |
| Reflection | *PLUS Num. obs: 5036 / % possible obs: 95.1 % / Num. measured all: 12467 / Rmerge(I) obs: 0.0737 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1LIN Resolution: 2.74→59.19 Å / Rfactor Rfree error: 0.017 / Data cutoff high absF: 1000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: GROUPED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.3 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.74→59.19 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.74→2.82 Å / Rfactor Rfree error: 0.098 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 2 / Rfactor Rfree: 0.2654 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
