 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-7540: Cryo-EM map of an HIV-1 reverse transcriptase initiation complex ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-7540 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM map of an HIV-1 reverse transcriptase initiation complex in magnesium chloride imaging buffer | |||||||||
 Map data Map data | HIV-1 RTIC core | |||||||||
 Sample Sample |
| |||||||||
| Biological species |   Human immunodeficiency virus 1 / Human immunodeficiency virus 1 /  Human (human) Human (human) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 8.2 Å | |||||||||
 Authors Authors | Larsen KP / Chen DH / Puglisi JD / Puglisi EV | |||||||||
 Citation Citation | Journal: Nature / Year: 2018 Title: Architecture of an HIV-1 reverse transcriptase initiation complex. Authors: Kevin P Larsen / Yamuna Kalyani Mathiharan / Kalli Kappel / Aaron T Coey / Dong-Hua Chen / Daniel Barrero / Lauren Madigan / Joseph D Puglisi / Georgios Skiniotis / Elisabetta Viani Puglisi /  Abstract: Reverse transcription of the HIV-1 RNA genome into double-stranded DNA is a central step in viral infection and a common target of antiretroviral drugs . The reaction is catalysed by viral reverse ...Reverse transcription of the HIV-1 RNA genome into double-stranded DNA is a central step in viral infection and a common target of antiretroviral drugs . The reaction is catalysed by viral reverse transcriptase (RT) that is packaged in an infectious virion with two copies of viral genomic RNA each bound to host lysine 3 transfer RNA (tRNA), which acts as a primer for initiation of reverse transcription. Upon viral entry into cells, initiation is slow and non-processive compared to elongation. Despite extensive efforts, the structural basis of RT function during initiation has remained a mystery. Here we use cryo-electron microscopy to determine a three-dimensional structure of an HIV-1 RT initiation complex. In our structure, RT is in an inactive polymerase conformation with open fingers and thumb and with the nucleic acid primer-template complex shifted away from the active site. The primer binding site (PBS) helix formed between tRNA and HIV-1 RNA lies in the cleft of RT and is extended by additional pairing interactions. The 5' end of the tRNA refolds and stacks on the PBS to create a long helical structure, while the remaining viral RNA forms two helical stems positioned above the RT active site, with a linker that connects these helices to the RNase H region of the PBS. Our results illustrate how RNA structure in the initiation complex alters RT conformation to decrease activity, highlighting a potential target for drug action. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_7540.map.gz | 58.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-7540-v30.xmlemd-7540.xml | 15.8 KB 15.8 KB | Display Display | EMDB header |
| Images | 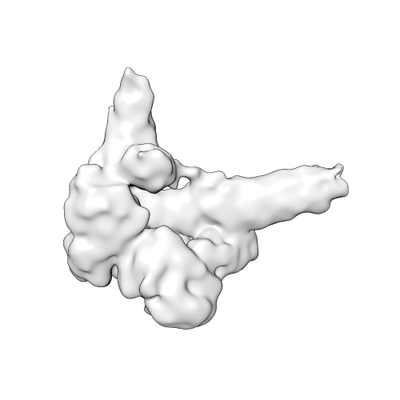 emd_7540.png emd_7540.png | 26.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-7540ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7540 http://ftp.pdbj.org/pub/emdb/structures/EMD-7540ftp://ftp.pdbj.org/pub/emdb/structures/EMD-7540 | HTTPS FTP |
-Related structure data











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_7540.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | HIV-1 RTIC core | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.286 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : HIV-1 reverse transcriptase initiation complex
| Entire | Name: HIV-1 reverse transcriptase initiation complex |
|---|---|
| Components |
|
-Supramolecule #1: HIV-1 reverse transcriptase initiation complex
| Supramolecule | Name: HIV-1 reverse transcriptase initiation complex / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#4 Details: Alternate buffer conditions containing magnesium chloride |
|---|---|
| Molecular weight | Theoretical: 33 KDa |
-Supramolecule #2: HIV-1 reverse transcriptase
| Supramolecule | Name: HIV-1 reverse transcriptase / type: complex / ID: 2 / Parent: 1 / Macromolecule list: #1-#2 Details: A cysteine mutation for crosslinking was introduced into helix H of p66 (Q258C). The protein used in this study also had the C280S mutation, introduced in prior structural work, and the ...Details: A cysteine mutation for crosslinking was introduced into helix H of p66 (Q258C). The protein used in this study also had the C280S mutation, introduced in prior structural work, and the E478Q mutation, introduced to eliminate RNase H activity. |
|---|---|
| Source (natural) | Organism: Human immunodeficiency virus 1 |
| Recombinant expression | Organism:  |
-Supramolecule #3: tRNA lysine3 primer
| Supramolecule | Name: tRNA lysine3 primer / type: complex / ID: 3 / Parent: 1 / Macromolecule list: #3 Details: Chemically synthesized and extended tRNA lysine3 primer. Modified nucleotide containing a N2-cystamine was placed at position 71. The tRNA primer has been extended by one ddCTP, bringing its ...Details: Chemically synthesized and extended tRNA lysine3 primer. Modified nucleotide containing a N2-cystamine was placed at position 71. The tRNA primer has been extended by one ddCTP, bringing its total length in the full complex to 77 nucleotides. |
|---|---|
| Source (natural) | Organism: Human (human) |
-Supramolecule #4: HIV-1 RNA genome fragment
| Supramolecule | Name: HIV-1 RNA genome fragment / type: complex / ID: 4 / Parent: 1 / Macromolecule list: #4 Details: HIV-1 RNA genome fragment of 101 nucleotides in length. Contains the primer binding site (PBS), primer activation signal (PAS), A-rich loop, and C-rich region. Generated via T7 transcription and PAGE purified. |
|---|---|
| Source (natural) | Organism: Human immunodeficiency virus 1 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4.5 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
Details: Beta-OG was added just prior to freezing. | |||||||||||||||
| Grid | Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: LACEY / Pretreatment - Type: GLOW DISCHARGE | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 85 % / Chamber temperature: 292 K / Instrument: LEICA EM GP Details: Blotted for 2.8 sec before plunging into liquid ethane.. | |||||||||||||||
| Details | Sample was monodisperse. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Frames/image: 2-60 / Number real images: 898 / Average exposure time: 12.0 sec. / Average electron dose: 75.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 38880 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.26 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 2.0 µm / Nominal magnification: 29000 |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
