 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of S. Mansoni p97 bound to CB-5083 | ||||||||||||||||||
 Map data Map data | |||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | AAA-ATPase / Endoplasmic Reticulum / Hexamer / Inhibitor / CHAPERONE | ||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationmitotic spindle disassembly / VCP-NPL4-UFD1 AAA ATPase complex / vesicle-fusing ATPase / retrograde protein transport, ER to cytosol / autophagosome maturation / polyubiquitin modification-dependent protein binding / ATP hydrolysis activity / ATP binding / nucleus / cytosol Similarity search - Function | ||||||||||||||||||
| Biological species |  | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.85 Å | ||||||||||||||||||
 Authors Authors | Stephens DR / Han Y / Chen Z / Collins JJ / Fung HYJ | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: Proc Natl Acad Sci U S A / Year: 2025 Title: A genome-scale drug discovery pipeline uncovers therapeutic targets and a unique p97 allosteric binding site in . Authors: Dylon R Stephens / Ho Yee Joyce Fung / Yan Han / Jue Liang / Zhe Chen / Joseph Ready / James J Collins / Abstract: Schistosomes are parasitic flatworms that infect more than 200 million people globally. However, there is a shortage of molecular tools that enable the discovery of potential drug targets within ...Schistosomes are parasitic flatworms that infect more than 200 million people globally. However, there is a shortage of molecular tools that enable the discovery of potential drug targets within schistosomes. Thus, praziquantel has remained the frontline treatment for schistosomiasis despite known liabilities. Here, we have conducted a genome-wide study in using the human druggable genome as a bioinformatic template to identify essential genes within schistosomes bearing similarity to catalogued drug targets. Then, we assessed these candidate targets in silico using a set of unbiased criteria to determine which possess ideal characteristics for a ready-made drug discovery campaign. Following this prioritization, we pursued a parasite p97 ortholog as a bona-fide drug target for the development of therapeutics to treat schistosomiasis. From this effort, we identified a covalent inhibitor series that kills schistosomes through an on-target killing mechanism by disrupting the ubiquitin proteasome system. Fascinatingly, these inhibitors induce a conformational change in the conserved D2 domain P-loop of schistosome p97 upon modification of Cys519. This conformational change reveals an allosteric binding site adjacent to the D2 domain active site reminiscent of the "DFG" flip in protein kinases. This allosteric binding site can potentially be utilized to generate new classes of species-selective p97 inhibitors. Furthermore, these studies provide a resource for the development of alternative therapeutics for schistosomiasis and a workflow to identify potential drug targets in similar systems with few available molecular tools. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_70961.map.gz | 74.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-70961-v30.xmlemd-70961.xml | 20.5 KB 20.5 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_70961_fsc.xml | 9.2 KB | Display | FSC data file |
| Images |  emd_70961.png emd_70961.png | 95.7 KB | ||
| Filedesc metadata | emd-70961.cif.gz | 7.1 KB | ||
| Others | emd_70961_half_map_1.map.gzemd_70961_half_map_2.map.gz | 77.4 MB 77.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-70961ftp://ftp.pdbj.org/pub/emdb/structures/EMD-70961 http://ftp.pdbj.org/pub/emdb/structures/EMD-70961ftp://ftp.pdbj.org/pub/emdb/structures/EMD-70961 | HTTPS FTP |
-Related structure data
| Related structure data |  9ox9MC  9p00C  9p01C  9p02C  9p07C M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |




-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |




-Map
| File | Download / File: emd_70961.map.gz / Format: CCP4 / Size: 83.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.0375 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: #2
| File | emd_70961_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: #1
| File | emd_70961_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : p97 Homohexamer bound to inhibitor CB-5083
| Entire | Name: p97 Homohexamer bound to inhibitor CB-5083 |
|---|---|
| Components |
|
-Supramolecule #1: p97 Homohexamer bound to inhibitor CB-5083
| Supramolecule | Name: p97 Homohexamer bound to inhibitor CB-5083 / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: vesicle-fusing ATPase
| Macromolecule | Name: vesicle-fusing ATPase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO / EC number: vesicle-fusing ATPase |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 92.755594 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MGSSHHHHHH SSGLVPRGSH MASMTGGQQM GRGSEFMCAL NANPSNDPSS GEKVKFHRLI VDEPVKDDNS VVYLSQAKMD SMNLFRGDT VLVKGKKRKE TVCVAIVDES CPDDKIRLNR CIRSNLRVKP GDIISIKSLP DILYGKRIHV LPIDDTIVGL T GNLYEAFL ...String: MGSSHHHHHH SSGLVPRGSH MASMTGGQQM GRGSEFMCAL NANPSNDPSS GEKVKFHRLI VDEPVKDDNS VVYLSQAKMD SMNLFRGDT VLVKGKKRKE TVCVAIVDES CPDDKIRLNR CIRSNLRVKP GDIISIKSLP DILYGKRIHV LPIDDTIVGL T GNLYEAFL KPYFLAAYRP VHKGDIFIVR GGMRAVEFKV IETDPSPYCI VSPDTTIHTE GDPVKREDEE EKLNEIGYDD IG GCRKQLA QIKEMVELPL RHPQLFKAIG VKPPRGILLY GPPGTGKTLV ARAVANESGS FFFLINGPEI MSKLAGESES NLR KAFEEA EKNAPAIIFI DELDAIAPKR EKTHGEVERR IVSQLLTLMD GLKQRSHVIV MAATNRPNSV DPALRRFGRF DREI EIGIP DSIGRLEILR IHTRNIRLAE DVELEKIANE AHGHVGADLA SLCSEAALQQ IRNKMNLIDL EDDTIDAEVL NSLAV TMDD FRWALGKSNP SALRETTVEV PNVTWDDIGG LENVKRELQE LVQYPVEHPD KFLKFGMTPS KGVLFYGPPG CGKTLL AKA IANECQANFI SIKGPELLTM WFGESEANVR DIFDKARQAA PCVLFFDELD SIAKARGGSV GDAGGAADRV INQLLTE MD GMSAKKNVFI IGATNRPDII DGAILRPGRL DQLIYIPLPD EASRVNILKA NLRKSPIARD VDINFLAKAT QGFSGADL T EICQRACKQA IRESIEAEIR AESEKKNKPN AMEDDFDPVP EITRRHFEEA MRFARRSVTE NDVRKYEMFA QTLQQSRGI GNNFRFPGSD GSGIPTSTGG QGGGGSVYGS QNDAEDLYN UniProtKB: vesicle-fusing ATPase |
-Macromolecule #2: 1-[4-(benzylamino)-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-2-yl]-2-...
| Macromolecule | Name: 1-[4-(benzylamino)-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-2-yl]-2-methyl-1H-indole-4-carboxamide type: ligand / ID: 2 / Number of copies: 6 / Formula: JDP |
|---|---|
| Molecular weight | Theoretical: 413.472 Da |
| Chemical component information | 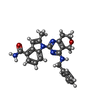 ChemComp-JDP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL |
|---|---|
| Buffer | pH: 7.4 Details: 20mM Tris (pH 7.4), 180mM NaCl, 5mM MgCl2, and 1mM tris(2-carboxyethyl)phosphine (TCEP) |
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 80 sec. |
| Vitrification | Cryogen name: ETHANE / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 10 eV |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Average electron dose: 62.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.4 µm / Nominal defocus min: 1.0 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Details | Model built by ModelAngelo for one chain. Other chains were manually fitted in Chimera and merged. |
|---|---|
| Refinement | Protocol: AB INITIO MODEL |
| Output model | PDB-9ox9: |
