 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-6284 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Yeast V-ATPase state 1 | |||||||||
 Map data Map data | Yeast V-ATPase state 1 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | V-ATPase / V-type ATPase / vacuolar-type ATPase / yeast / Saccharomyces cerevisiae / hydrolase | |||||||||
| Function / homology |  Function and homology information Function and homology informationvacuole-mitochondrion membrane contact site / protein localization to vacuolar membrane / cellular response to alkaline pH / Insulin receptor recycling / Transferrin endocytosis and recycling / polyphosphate metabolic process / ROS and RNS production in phagocytes / Golgi lumen acidification / Amino acids regulate mTORC1 / proteasome storage granule assembly ...vacuole-mitochondrion membrane contact site / protein localization to vacuolar membrane / cellular response to alkaline pH / Insulin receptor recycling / Transferrin endocytosis and recycling / polyphosphate metabolic process / ROS and RNS production in phagocytes / Golgi lumen acidification / Amino acids regulate mTORC1 / proteasome storage granule assembly / vacuolar transport / vacuolar proton-transporting V-type ATPase, V1 domain / vacuolar proton-transporting V-type ATPase, V0 domain / endosomal lumen acidification / vacuole organization / proton-transporting V-type ATPase complex / fungal-type vacuole / intron homing / protein targeting to vacuole / vacuolar proton-transporting V-type ATPase complex / intein-mediated protein splicing / pexophagy / cellular hyperosmotic response / vacuolar acidification / fungal-type vacuole membrane / phosphatidylinositol-3,5-bisphosphate binding / proton transmembrane transporter activity / proton-transporting ATPase activity, rotational mechanism / intracellular copper ion homeostasis / H+-transporting two-sector ATPase / ATP metabolic process / Neutrophil degranulation / proton transmembrane transport / transmembrane transport / endocytosis / intracellular calcium ion homeostasis / cytoplasmic stress granule / ATPase binding / protein-containing complex assembly / endonuclease activity / Hydrolases; Acting on ester bonds / intracellular iron ion homeostasis / membrane raft / Golgi membrane / hydrolase activity / mRNA binding / ATP hydrolysis activity / DNA binding / ATP binding / membrane / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.9 Å | |||||||||
 Authors Authors | Zhao J / Benlekbir S / Rubinstein JL | |||||||||
 Citation Citation | Journal: Nature / Year: 2015 Title: Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Authors: Jianhua Zhao / Samir Benlekbir / John L Rubinstein /  Abstract: Eukaryotic vacuolar H(+)-ATPases (V-ATPases) are rotary enzymes that use energy from hydrolysis of ATP to ADP to pump protons across membranes and control the pH of many intracellular compartments. ...Eukaryotic vacuolar H(+)-ATPases (V-ATPases) are rotary enzymes that use energy from hydrolysis of ATP to ADP to pump protons across membranes and control the pH of many intracellular compartments. ATP hydrolysis in the soluble catalytic region of the enzyme is coupled to proton translocation through the membrane-bound region by rotation of a central rotor subcomplex, with peripheral stalks preventing the entire membrane-bound region from turning with the rotor. The eukaryotic V-ATPase is the most complex rotary ATPase: it has three peripheral stalks, a hetero-oligomeric proton-conducting proteolipid ring, several subunits not found in other rotary ATPases, and is regulated by reversible dissociation of its catalytic and proton-conducting regions. Studies of ATP synthases, V-ATPases, and bacterial/archaeal V/A-ATPases have suggested that flexibility is necessary for the catalytic mechanism of rotary ATPases, but the structures of different rotational states have never been observed experimentally. Here we use electron cryomicroscopy to obtain structures for three rotational states of the V-ATPase from the yeast Saccharomyces cerevisiae. The resulting series of structures shows ten proteolipid subunits in the c-ring, setting the ATP:H(+) ratio for proton pumping by the V-ATPase at 3:10, and reveals long and highly tilted transmembrane α-helices in the a-subunit that interact with the c-ring. The three different maps reveal the conformational changes that occur to couple rotation in the symmetry-mismatched soluble catalytic region to the membrane-bound proton-translocating region. Almost all of the subunits of the enzyme undergo conformational changes during the transitions between these three rotational states. The structures of these states provide direct evidence that deformation during rotation enables the smooth transmission of power through rotary ATPases. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_6284.map.gz | 58.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-6284-v30.xmlemd-6284.xml | 13.7 KB 13.7 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_6284_fsc.xml | 10.9 KB | Display | FSC data file |
| Images | 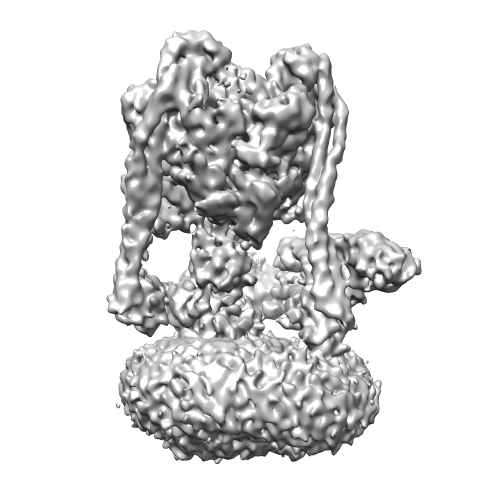 emd_6284.png emd_6284.png | 127.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-6284ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6284 http://ftp.pdbj.org/pub/emdb/structures/EMD-6284ftp://ftp.pdbj.org/pub/emdb/structures/EMD-6284 | HTTPS FTP |
-Related structure data
| Related structure data |  3j9tMC  6285C  6286C  3j9uC  3j9vC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |












-Map
| File | Download / File: emd_6284.map.gz / Format: CCP4 / Size: 62.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Yeast V-ATPase state 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.45 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
- Sample components
Sample components
-Entire : Vacuolar-type ATPase
| Entire | Name: Vacuolar-type ATPase |
|---|---|
| Components |
|
-Supramolecule #1000: Vacuolar-type ATPase
| Supramolecule | Name: Vacuolar-type ATPase / type: sample / ID: 1000 / Details: Detergent solubilized protein complex Oligomeric state: A3B3CDE3FG3HadcXc'Yc''Z where X, Y, and Z indicate unknown stoichiometry and X+Y+Z=10 Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 900 KDa / Theoretical: 900 KDa / Method: SDS-PAGE, size exclusion chromatography |
-Macromolecule #1: Vacuolar-type ATPase
| Macromolecule | Name: Vacuolar-type ATPase / type: protein_or_peptide / ID: 1 / Name.synonym: V-ATPase, V-type ATPase / Details: Detergent-solubilized protein complex / Number of copies: 1 / Oligomeric state: monomer / Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 900 KDa / Theoretical: 900 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 10 mg/mL |
|---|---|
| Buffer | pH: 7.4 Details: 50 mM Tris-HCl, 150 mM NaCl, 0.02% w/v dodecylmaltoside |
| Grid | Details: Homemade holey carbon on 400 square mesh Cu/Rh grid, glow-discharged 2 mins |
| Vitrification | Cryogen name: ETHANE-PROPANE MIXTURE / Chamber humidity: 100 % / Chamber temperature: 77 K / Instrument: FEI VITROBOT MARK III / Method: Blot for 23 seconds before freezing. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Alignment procedure | Legacy - Astigmatism: Manually corrected by inspecting FFT. |
| Details | K2 Summit in counting mode, 2 frames/s for 15 s |
| Date | Oct 19, 2013 |
| Image recording | Category: CCD / Film or detector model: GATAN K2 (4k x 4k) / Number real images: 3685 / Average electron dose: 30 e/Å2 / Bits/pixel: 32 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 34483 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 7.0 µm / Nominal defocus min: 1.5 µm / Nominal magnification: 34483 |
| Sample stage | Specimen holder model: GATAN LIQUID NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| Details | Particles automatically selected using TMaCS and processed in Relion. |
|---|---|
| CTF correction | Details: Each particle |
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 6.9 Å / Resolution method: OTHER / Software - Name: Relion / Number images used: 50503 |
| FSC plot (resolution estimation) |  |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-3j9t: |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-3j9t: |

-Atomic model buiding 3
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-3j9t: |

-Atomic model buiding 4
| Initial model | PDB ID: Chain - #0 - Chain ID: E / Chain - #1 - Chain ID: G |
|---|---|
| Software | Name: Chimera, MDFF |
| Details | Rigid body fitting performed in Chimera first, followed by flexible fitting performed using Molecular Dynamics Flexible Fitting (MDFF). |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT |
| Output model | PDB-3j9t: |

