 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of Nap1 core | ||||||||||||||||||
 Map data Map data | Main map | ||||||||||||||||||
 Sample Sample |
| ||||||||||||||||||
 Keywords Keywords | Histone / Chaperone | ||||||||||||||||||
| Function / homology | :  Function and homology information Function and homology information | ||||||||||||||||||
| Biological species |  | ||||||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.2 Å | ||||||||||||||||||
 Authors Authors | Jiou J / Fung HYJ / Chook YM | ||||||||||||||||||
| Funding support |  United States, 5 items United States, 5 items
| ||||||||||||||||||
 Citation Citation | Journal: bioRxiv / Year: 2024 Title: Nap1 and Kap114 co-chaperone H2A-H2B and facilitate targeted histone release in the nucleus. Authors: Ho Yee Joyce Fung / Ashley B Neisman / Natalia E Bernardes / Jenny Jiou / Yuh Min Chook Abstract: Core histones are synthesized and processed in the cytoplasm before transport into the nucleus for assembly into nucleosomes; however, they must also be chaperoned as free histones are toxic. The ...Core histones are synthesized and processed in the cytoplasm before transport into the nucleus for assembly into nucleosomes; however, they must also be chaperoned as free histones are toxic. The importin Kap114 binds and transports histone H2A-H2B into the yeast nucleus, where RanGTP facilitates H2A-H2B release. Kap114 and H2A-H2B also bind the Nap1 histone chaperone, which is found in both the cytoplasm and the nucleus, but how Nap1 and Kap114 cooperate in H2A-H2B processing and nucleosome assembly has been unclear. To understand these mechanisms, we used biochemical and structural analyses to reveal how Nap1, Kap114, H2A-H2B and RanGTP interact. We show that Kap114, H2A-H2B and a Nap1 dimer (Nap1 ) assemble into a 1:1:1 ternary complex. Cryogenic electron microscopy revealed two distinct Kap114/Nap1 /H2A-H2B structures: one of H2A-H2B sandwiched between Nap1 and Kap114, and another in which Nap1 bound to the Kap114·H2A-H2B complex without contacting H2A-H2B. Another Nap1 ·H2A-H2B·Kap114·Ran structure reveals the nuclear complex. Mutagenesis revealed shared critical interfaces in all three structures. Consistent with structural findings, DNA competition experiments demonstrated that Kap114 and Nap1 together chaperone H2A-H2B better than either protein alone. When RanGTP is present, Kap114's chaperoning activity diminishes. However, the presence of Nap1 within the Nap1 ·H2A-H2B·Kap114·Ran quaternary complex restores its ability to chaperone H2A-H2B. This complex effectively deposits H2A-H2B into nucleosomes. Together, these findings suggest that Kap114 and Nap12 provide a sheltered path from cytoplasm to nucleus, facilitating the transfer of H2A-H2B from Kap114 to Nap1 , ultimately directing its specific deposition into nucleosomes. | ||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_44095.map.gz | 41.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-44095-v30.xmlemd-44095.xml | 19.4 KB 19.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_44095_fsc.xml | 12.8 KB | Display | FSC data file |
| Images |  emd_44095.png emd_44095.png | 47.6 KB | ||
| Filedesc metadata | emd-44095.cif.gz | 6.3 KB | ||
| Others | emd_44095_half_map_1.map.gzemd_44095_half_map_2.map.gz | 77.6 MB 77.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-44095ftp://ftp.pdbj.org/pub/emdb/structures/EMD-44095 http://ftp.pdbj.org/pub/emdb/structures/EMD-44095ftp://ftp.pdbj.org/pub/emdb/structures/EMD-44095 | HTTPS FTP |
-Related structure data
| Related structure data |  9b23MC  9b31C  9b3fC  9b3iC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_44095.map.gz / Format: CCP4 / Size: 83.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Main map | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.83 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: Half B
| File | emd_44095_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
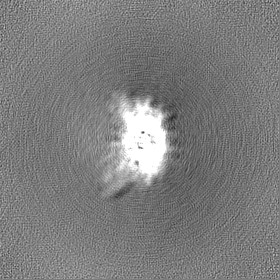
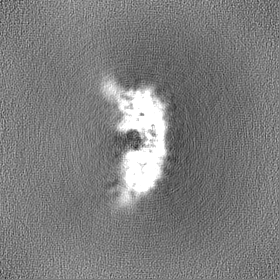






-Half map: Half A
| File | emd_44095_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Complex of Kap114 bound to Nap1 and histone H2A-H2B
| Entire | Name: Complex of Kap114 bound to Nap1 and histone H2A-H2B |
|---|---|
| Components |
|
-Supramolecule #1: Complex of Kap114 bound to Nap1 and histone H2A-H2B
| Supramolecule | Name: Complex of Kap114 bound to Nap1 and histone H2A-H2B / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all / Details: crosslinked sample. |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 72 KDa |
-Macromolecule #1: NAP1 isoform 1
| Macromolecule | Name: NAP1 isoform 1 / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 36.182355 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MGSSHHHHHH SSGLVPRGSH MLGSLVGQDS GYVGGLPKNV KEKLLSLKTL QSELFEVEKE FQVEMFELEN KFLQKYKPIW EQRSRIISG QEQPKPEQIA KGQEIVESLN ETELLVDEEE KAQNDSEEEQ VKGIPSFWLT ALENLPIVCD TITDRDAEVL E YLQDIGLE ...String: MGSSHHHHHH SSGLVPRGSH MLGSLVGQDS GYVGGLPKNV KEKLLSLKTL QSELFEVEKE FQVEMFELEN KFLQKYKPIW EQRSRIISG QEQPKPEQIA KGQEIVESLN ETELLVDEEE KAQNDSEEEQ VKGIPSFWLT ALENLPIVCD TITDRDAEVL E YLQDIGLE YLTDGRPGFK LLFRFDSSAN PFFTNDILCK TYFYQKELGY SGDFIYDHAE GCEISWKDNA HNVTVDLEMR KQ RNKTTKQ VRTIEKITPI ESFFNFFDPP KIQNEDQDEE LEEDLEERLA LDYSIGEQLK DKLIPRAVDW FTGAAL UniProtKB: UNIPROTKB: A0A8H4BY55 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.2 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.4 Component:
| |||||||||||||||
| Grid | Model: Quantifoil / Material: COPPER / Mesh: 300 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 280 K / Instrument: FEI VITROBOT MARK IV | |||||||||||||||
| Details | Crosslinked sample. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Number grids imaged: 1 / Number real images: 1080 / Average exposure time: 5.4 sec. / Average electron dose: 59.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 2.4 µm / Nominal defocus min: 0.9 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | Chain - Source name: AlphaFold / Chain - Initial model type: in silico model Details: AlphaFold Multimer was used to generate initial model. |
|---|---|
| Refinement | Protocol: OTHER |
| Output model | PDB-9b23: |
