Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4347 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
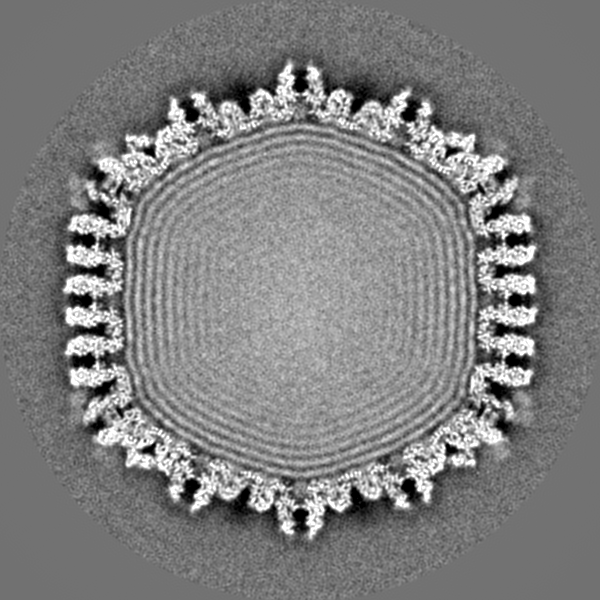
| Title | Structure of the herpes-simplex virus portal-vertex | |||||||||
 Map data Map data | Reconstruction of the herpes simplex virus type 1 virion, with relaxed (C5) symmetry. The map is sharpened. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Herpes simplex virus (type 1 / strain 17) Herpes simplex virus (type 1 / strain 17) | |||||||||
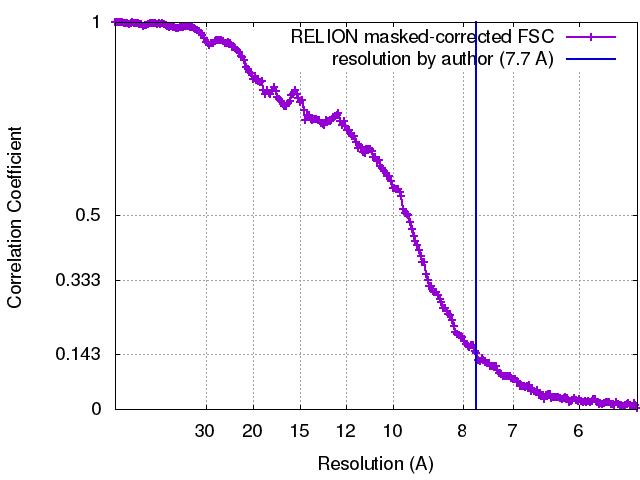
| Method | single particle reconstruction / cryo EM / Resolution: 7.7 Å | |||||||||
 Authors Authors | McElwee M / Vijayakrishnan S / Rixon FJ / Bhella D | |||||||||
 Citation Citation | Journal: PLoS Biol / Year: 2018 Title: Structure of the herpes simplex virus portal-vertex. Authors: Marion McElwee / Swetha Vijayakrishnan / Frazer Rixon / David Bhella /  Abstract: Herpesviruses include many important human pathogens such as herpes simplex virus, cytomegalovirus, varicella-zoster virus, and the oncogenic Epstein-Barr virus and Kaposi sarcoma-associated ...Herpesviruses include many important human pathogens such as herpes simplex virus, cytomegalovirus, varicella-zoster virus, and the oncogenic Epstein-Barr virus and Kaposi sarcoma-associated herpesvirus. Herpes virions contain a large icosahedral capsid that has a portal at a unique 5-fold vertex, similar to that seen in the tailed bacteriophages. The portal is a molecular motor through which the viral genome enters the capsid during virion morphogenesis. The genome also exits the capsid through the portal-vertex when it is injected through the nuclear pore into the nucleus of a new host cell to initiate infection. Structural investigations of the herpesvirus portal-vertex have proven challenging, owing to the small size of the tail-like portal-vertex-associated tegument (PVAT) and the presence of the tegument layer that lays between the nucleocapsid and the viral envelope, obscuring the view of the portal-vertex. Here, we show the structure of the herpes simplex virus portal-vertex at subnanometer resolution, solved by electron cryomicroscopy (cryoEM) and single-particle 3D reconstruction. This led to a number of new discoveries, including the presence of two previously unknown portal-associated structures that occupy the sites normally taken by the penton and the Ta triplex. Our data revealed that the PVAT is composed of 10 copies of the C-terminal domain of pUL25, which are uniquely arranged as two tiers of star-shaped density. Our 3D reconstruction of the portal-vertex also shows that one end of the viral genome extends outside the portal in the manner described for some bacteriophages but not previously seen in any eukaryote viruses. Finally, we show that the viral genome is consistently packed in a highly ordered left-handed spool to form concentric shells of DNA. Our data provide new insights into the structure of a molecular machine critical to the biology of an important class of human pathogens. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
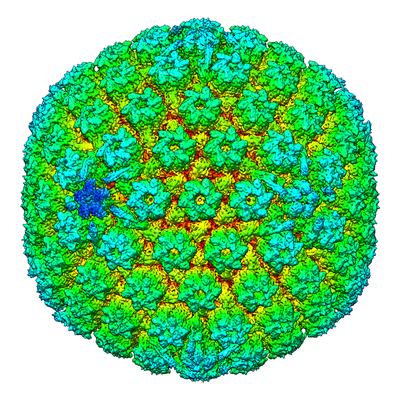
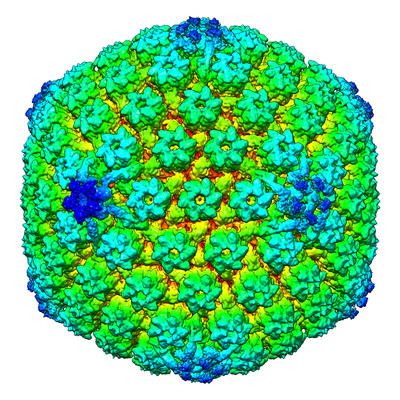
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4347.map.gz | 747.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4347-v30.xmlemd-4347.xml | 16 KB 16 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_4347_fsc.xml | 21.2 KB | Display | FSC data file |
| Images |  emd_4347_1.png emd_4347_1.png emd_4347_2.png emd_4347_2.png | 267 KB 253.4 KB | ||
| Others | emd_4347_additional.map.gz | 734.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4347ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4347 http://ftp.pdbj.org/pub/emdb/structures/EMD-4347ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4347 | HTTPS FTP |
-Related structure data
| Similar structure data | |
|---|---|
| EM raw data | EMPIAR-10189 (Title: Structure of the herpes-simplex virus portal-vertex / Data size: 238.6 Data #1: Motion corrected micrographs of herpes simplex virus type 1 virions [micrographs - single frame]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_4347.map.gz / Format: CCP4 / Size: 824 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of the herpes simplex virus type 1 virion, with relaxed (C5) symmetry. The map is sharpened. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
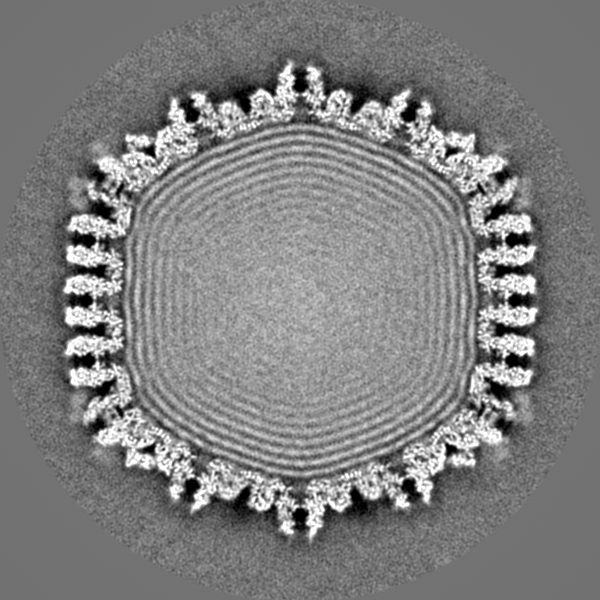
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.67 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Additional map: Reconstruction of the herpes simplex virus type 1...
| File | emd_4347_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
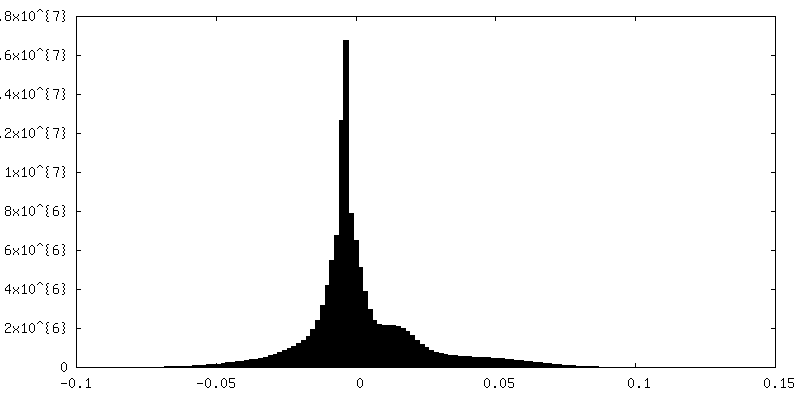
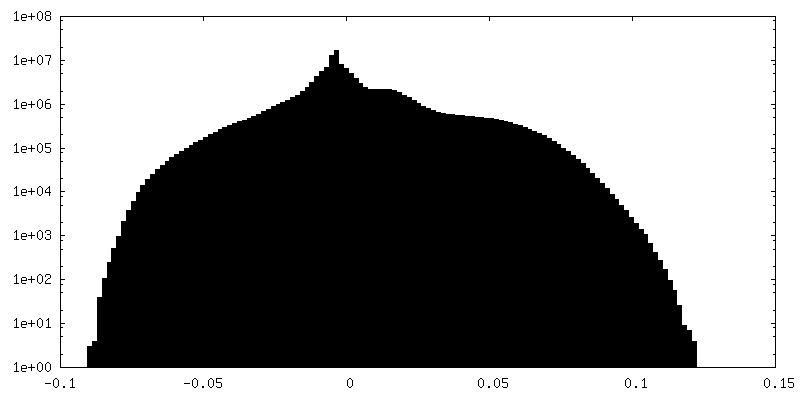
| Annotation | Reconstruction of the herpes simplex virus type 1 virion, with relaxed (C5) symmetry. The map is NOT sharpened. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : Herpes simplex virus (type 1 / strain 17)
| Entire | Name: Herpes simplex virus (type 1 / strain 17) |
|---|---|
| Components |
|
-Supramolecule #1: Herpes simplex virus (type 1 / strain 17)
| Supramolecule | Name: Herpes simplex virus (type 1 / strain 17) / type: virus / ID: 1 / Parent: 0 / Details: Virus was propagated in BHK cells / NCBI-ID: 10299 / Sci species name: Herpes simplex virus (type 1 / strain 17) / Virus type: VIRION / Virus isolate: STRAIN / Virus enveloped: Yes / Virus empty: No |
|---|---|
| Host (natural) | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Virus shell | Shell ID: 1 / Name: Capsid / Diameter: 1250.0 Å / T number (triangulation number): 16 |
-Supramolecule #2: Portal
| Supramolecule | Name: Portal / type: complex / ID: 2 / Parent: 1 / Details: Dodecameric portal comprised of the pUL6 protein |
|---|---|
| Source (natural) | Organism: Herpes simplex virus (type 1 / strain 17) |
-Supramolecule #3: Portal-vertex associated tegument protein
| Supramolecule | Name: Portal-vertex associated tegument protein / type: complex / ID: 3 / Parent: 1 Details: portal associated tail-like density comprised of ten copies of the pUL25 protein |
|---|---|
| Source (natural) | Organism: Herpes simplex virus (type 1 / strain 17) |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1 mg/mL |
|---|---|
| Buffer | pH: 7.2 / Details: Phosphate buffered saline |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
| Details | Purified enveloped virions |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: INTEGRATING / Number grids imaged: 1 / Number real images: 3702 / Average exposure time: 12.0 sec. / Average electron dose: 78.0 e/Å2 Details: Images were acquired as 40 fractions per micrograph at 1.78 angstroms/pixel sampling. |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 81000 |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |