 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Cryo-EM structure of designed Influenza HA binder, HA_20, bound to Influenza HA (Strain: Iowa43) | |||||||||
 マップデータ マップデータ | unsharpened | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | flu / influenza / hemagglutinin / HA / Iowa43 / HA_20 / DE NOVO PROTEIN / minibinder / binder / designed protein / fusion protein / glycoprotein / DE NOVO PROTEIN-Viral Protein complex | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報viral budding from plasma membrane / clathrin-dependent endocytosis of virus by host cell / host cell surface receptor binding / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / virion attachment to host cell / host cell plasma membrane / virion membrane / membrane 類似検索 - 分子機能 | |||||||||
| 生物種 |   Influenza A virus (A型インフルエンザウイルス) / unidentified (未定義) Influenza A virus (A型インフルエンザウイルス) / unidentified (未定義) | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.93 Å | |||||||||
 データ登録者 データ登録者 | Borst AJ / Bennett NR | |||||||||
| 資金援助 |  米国, 1件 米国, 1件
| |||||||||
 引用 引用 | ジャーナル: Nature / 年: 2023 タイトル: De novo design of protein structure and function with RFdiffusion. 著者: Joseph L Watson / David Juergens / Nathaniel R Bennett / Brian L Trippe / Jason Yim / Helen E Eisenach / Woody Ahern / Andrew J Borst / Robert J Ragotte / Lukas F Milles / Basile I M Wicky / ...著者: Joseph L Watson / David Juergens / Nathaniel R Bennett / Brian L Trippe / Jason Yim / Helen E Eisenach / Woody Ahern / Andrew J Borst / Robert J Ragotte / Lukas F Milles / Basile I M Wicky / Nikita Hanikel / Samuel J Pellock / Alexis Courbet / William Sheffler / Jue Wang / Preetham Venkatesh / Isaac Sappington / Susana Vázquez Torres / Anna Lauko / Valentin De Bortoli / Emile Mathieu / Sergey Ovchinnikov / Regina Barzilay / Tommi S Jaakkola / Frank DiMaio / Minkyung Baek / David Baker /    要旨: There has been considerable recent progress in designing new proteins using deep-learning methods. Despite this progress, a general deep-learning framework for protein design that enables solution of ...There has been considerable recent progress in designing new proteins using deep-learning methods. Despite this progress, a general deep-learning framework for protein design that enables solution of a wide range of design challenges, including de novo binder design and design of higher-order symmetric architectures, has yet to be described. Diffusion models have had considerable success in image and language generative modelling but limited success when applied to protein modelling, probably due to the complexity of protein backbone geometry and sequence-structure relationships. Here we show that by fine-tuning the RoseTTAFold structure prediction network on protein structure denoising tasks, we obtain a generative model of protein backbones that achieves outstanding performance on unconditional and topology-constrained protein monomer design, protein binder design, symmetric oligomer design, enzyme active site scaffolding and symmetric motif scaffolding for therapeutic and metal-binding protein design. We demonstrate the power and generality of the method, called RoseTTAFold diffusion (RFdiffusion), by experimentally characterizing the structures and functions of hundreds of designed symmetric assemblies, metal-binding proteins and protein binders. The accuracy of RFdiffusion is confirmed by the cryogenic electron microscopy structure of a designed binder in complex with influenza haemagglutinin that is nearly identical to the design model. In a manner analogous to networks that produce images from user-specified inputs, RFdiffusion enables the design of diverse functional proteins from simple molecular specifications. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 添付画像 |
|---|

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_40557.map.gz | 75 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-40557-v30.xmlemd-40557.xml | 21.7 KB 21.7 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_40557_fsc.xml | 11.2 KB | 表示 | FSCデータファイル |
| 画像 | 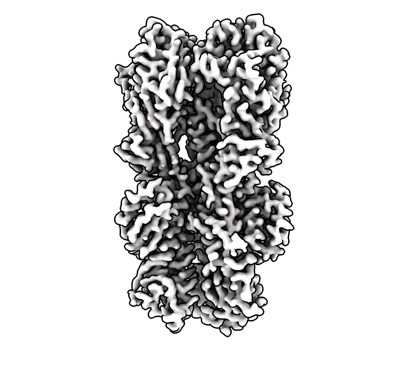 emd_40557.png emd_40557.png | 113.8 KB | ||
| その他 | emd_40557_additional_1.map.gzemd_40557_additional_2.map.gzemd_40557_half_map_1.map.gzemd_40557_half_map_2.map.gz | 125.2 MB 75 MB 138.7 MB 138.7 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-40557ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40557 http://ftp.pdbj.org/pub/emdb/structures/EMD-40557ftp://ftp.pdbj.org/pub/emdb/structures/EMD-40557 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_40557_validation.pdf.gz | 780.2 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_40557_full_validation.pdf.gz | 779.8 KB | 表示 | |
| XML形式データ | emd_40557_validation.xml.gz | 19.9 KB | 表示 | |
| CIF形式データ | emd_40557_validation.cif.gz | 25.9 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-40557ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-40557 | HTTPS FTP |
-関連構造データ

-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |






-マップ
| ファイル | ダウンロード / ファイル: emd_40557.map.gz / 形式: CCP4 / 大きさ: 149.9 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | unsharpened | ||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.83 Å | ||||||||||||||||||||||||||||||||||||
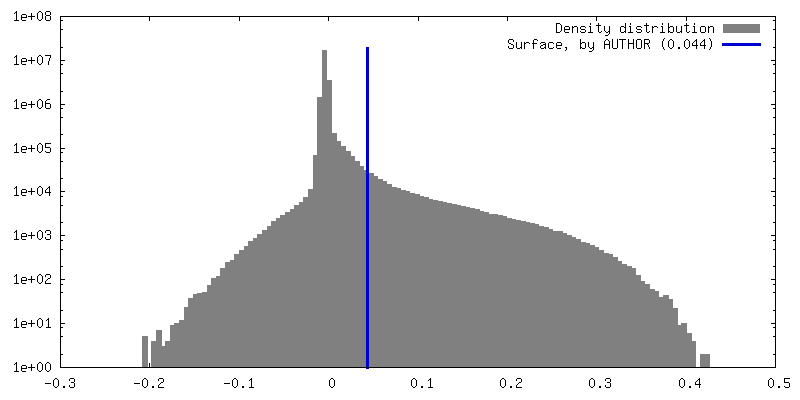
| 密度 |
| ||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-添付データ
-追加マップ: deepEMhancer
| ファイル | emd_40557_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | deepEMhancer | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |








-追加マップ: autosharpened
| ファイル | emd_40557_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | autosharpened | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |








-ハーフマップ: half map a
| ファイル | emd_40557_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half map a | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |








-ハーフマップ: half map b
| ファイル | emd_40557_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half map b | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |






- 試料の構成要素
試料の構成要素
-全体 : Influenza HA (Iowa43) bound to RFdiffusion designed minibinder, HA_20
| 全体 | 名称: Influenza HA (Iowa43) bound to RFdiffusion designed minibinder, HA_20 |
|---|---|
| 要素 |
|
-超分子 #1: Influenza HA (Iowa43) bound to RFdiffusion designed minibinder, HA_20
| 超分子 | 名称: Influenza HA (Iowa43) bound to RFdiffusion designed minibinder, HA_20 タイプ: complex / ID: 1 / 親要素: 0 / 含まれる分子: #1-#3 |
|---|---|
| 由来(天然) | 生物種: Influenza A virus (A型インフルエンザウイルス) |
-分子 #1: Hemagglutinin HA1 chain
| 分子 | 名称: Hemagglutinin HA1 chain / タイプ: protein_or_peptide / ID: 1 / コピー数: 3 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Influenza A virus (A型インフルエンザウイルス) |
| 分子量 | 理論値: 35.92334 KDa |
| 組換発現 | 生物種:  Homo sapiens (ヒト) Homo sapiens (ヒト) |
| 配列 | 文字列: DTICIGYHAN NSTDTVDTVL EKNVTVTHSV NLLEDSHNGK LCRLKGIAPL QLGKCNIAGW ILGNPECESL LSERSWSYIV ETPNSENGT CFPGDFIDYE ELREQLSSVS SFERFEIFSK ESSWPKHTTG GVTAACSHAG KSSFYRNLLW LTEKDGSYPN L NNSYVNKK ...文字列: DTICIGYHAN NSTDTVDTVL EKNVTVTHSV NLLEDSHNGK LCRLKGIAPL QLGKCNIAGW ILGNPECESL LSERSWSYIV ETPNSENGT CFPGDFIDYE ELREQLSSVS SFERFEIFSK ESSWPKHTTG GVTAACSHAG KSSFYRNLLW LTEKDGSYPN L NNSYVNKK GKEVLVLWGV HHPSNIKDQQ TLYQKENAYV SVVSSNYNRR FTPEIAERPK VRGQAGRINY YWTLLKPGDT IM FEANGNL IAPWYAFALS RGFGSGIITS NASMHECDTK CQTPQGAINS SLPFQNIHPI TIGECPKYVR STKLRMVTGL RNI P UniProtKB: Hemagglutinin |
-分子 #2: Hemagglutinin
| 分子 | 名称: Hemagglutinin / タイプ: protein_or_peptide / ID: 2 / コピー数: 3 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: Influenza A virus (A型インフルエンザウイルス) |
| 分子量 | 理論値: 26.623463 KDa |
| 組換発現 | 生物種: Homo sapiens (ヒト) |
| 配列 | 文字列: SIQSRGLFGA IAGFIEGGWT GMIDGWYGYH WQNEQGSGYA ADQKSTQNAI NGITNIVNSV IEKMNTQFTA VGKEFNNLEK RMENLNKKV DDGFLDIWTY NAELLVLLIN ERTLDFHDSN VKNLYEKVKN QLRNNAKEIG NGCFEFYHKC NNECMESVKN G TYDYPKYS ...文字列: SIQSRGLFGA IAGFIEGGWT GMIDGWYGYH WQNEQGSGYA ADQKSTQNAI NGITNIVNSV IEKMNTQFTA VGKEFNNLEK RMENLNKKV DDGFLDIWTY NAELLVLLIN ERTLDFHDSN VKNLYEKVKN QLRNNAKEIG NGCFEFYHKC NNECMESVKN G TYDYPKYS EESKLNREKI DGSGYIPEAP RDGQAYVRKD GEWVLLSTFL GSGLNDIFEA QKIEWHEGHH HHHH UniProtKB: Hemagglutinin |
-分子 #3: HA_20 minibinder (RFdiffusion-designed)
| 分子 | 名称: HA_20 minibinder (RFdiffusion-designed) / タイプ: protein_or_peptide / ID: 3 / コピー数: 3 / 光学異性体: LEVO |
|---|---|
| 由来(天然) | 生物種: unidentified (未定義) |
| 分子量 | 理論値: 7.391848 KDa |
| 組換発現 | 生物種:  |
| 配列 | 文字列: MEKEKELKEY AEKIKKEIGD IESVEVKDGK ILVKAKKITD KTVDAIMKLT VKAARLGFKV EVELV |
-分子 #6: 2-acetamido-2-deoxy-beta-D-glucopyranose
| 分子 | 名称: 2-acetamido-2-deoxy-beta-D-glucopyranose / タイプ: ligand / ID: 6 / コピー数: 12 / 式: NAG |
|---|---|
| 分子量 | 理論値: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 緩衝液 | pH: 7.5 |
|---|---|
| 凍結 | 凍結剤: ETHANE |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | TFS KRIOS |
|---|---|
| 撮影 | フィルム・検出器のモデル: GATAN K3 BIOQUANTUM (6k x 4k) 平均電子線量: 64.273 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / 最大 デフォーカス(公称値): 1.7 µm / 最小 デフォーカス(公称値): 0.8 µm |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |


