 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-3664: Structure of the Bacillus subtilis hibernating 100S ribosome reve... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-3664 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. | |||||||||
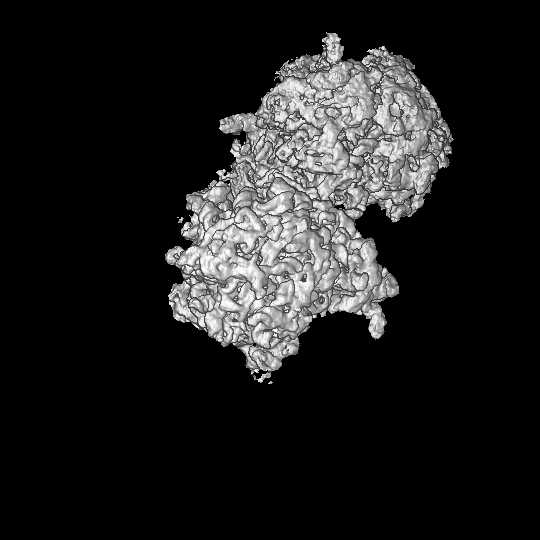
 Map data Map data | Cryo-EM structure of the Bacillus subtilis 100S ribosome, filtered and masked final map | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 6.2 Å | |||||||||
 Authors Authors | Beckert B / Abdelshahid M / Schafer H / Steinchen W / Arenz S / Berninghausen O / Beckmann R / Bange G / Turgay K / Wilson DN | |||||||||
 Citation Citation | Journal: EMBO J / Year: 2017 Title: Structure of the hibernating 100S ribosome reveals the basis for 70S dimerization. Authors: Bertrand Beckert / Maha Abdelshahid / Heinrich Schäfer / Wieland Steinchen / Stefan Arenz / Otto Berninghausen / Roland Beckmann / Gert Bange / Kürşad Turgay / Daniel N Wilson /  Abstract: Under stress conditions, such as nutrient deprivation, bacteria enter into a hibernation stage, which is characterized by the appearance of 100S ribosomal particles. In , dimerization of 70S ...Under stress conditions, such as nutrient deprivation, bacteria enter into a hibernation stage, which is characterized by the appearance of 100S ribosomal particles. In , dimerization of 70S ribosomes into 100S requires the action of the ribosome modulation factor (RMF) and the hibernation-promoting factor (HPF). Most other bacteria lack RMF and instead contain a long form HPF (LHPF), which is necessary and sufficient for 100S formation. While some structural information exists as to how RMF and HPF mediate formation of 100S (100S), structural insight into 100S formation by LHPF has so far been lacking. Here we present a cryo-EM structure of the hibernating 100S (100S), revealing that the C-terminal domain (CTD) of the LHPF occupies a site on the 30S platform distinct from RMF Moreover, unlike RMF, the HPF-CTD is directly involved in forming the dimer interface, thereby illustrating the divergent mechanisms by which 100S formation is mediated in the majority of bacteria that contain LHPF, compared to some γ-proteobacteria, such as . | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_3664.map.gz | 44.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-3664-v30.xmlemd-3664.xml | 16.9 KB 16.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_3664_fsc.xml | 17.3 KB | Display | FSC data file |
| Images | 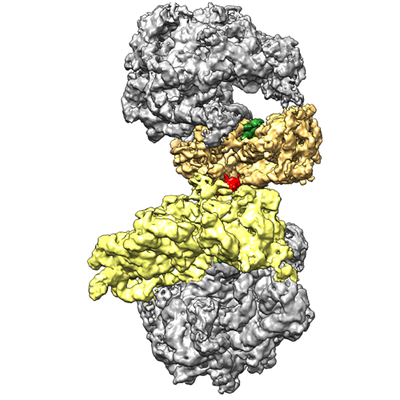 emd_3664.png emd_3664.png | 144.9 KB | ||
| Others | emd_3664_additional.map.gzemd_3664_additional_1.map.gzemd_3664_half_map_1.map.gzemd_3664_half_map_2.map.gz | 392.5 MB 392.5 MB 393.1 MB 393.1 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-3664ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3664 http://ftp.pdbj.org/pub/emdb/structures/EMD-3664ftp://ftp.pdbj.org/pub/emdb/structures/EMD-3664 | HTTPS FTP |
-Related structure data












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_3664.map.gz / Format: CCP4 / Size: 600.7 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of the Bacillus subtilis 100S ribosome, filtered and masked final map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.084 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Additional map: Cryo-EM structure of the Bacillus subtilis 100S ribosome,...
| File | emd_3664_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of the Bacillus subtilis 100S ribosome, filtered final map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Additional map: Cryo-EM structure of the Bacillus subtilis 100S ribosome,...
| File | emd_3664_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of the Bacillus subtilis 100S ribosome, filtered final map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Cryo-EM structure of the Bacillus subtilis 100S ribosome, half map
| File | emd_3664_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of the Bacillus subtilis 100S ribosome, half map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: Cryo-EM structure of the Bacillus subtilis 100S ribosome, half map
| File | emd_3664_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM structure of the Bacillus subtilis 100S ribosome, half map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : Structure of the Bacillus subtilis hibernating 100S ribosome reve...
| Entire | Name: Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. |
|---|---|
| Components |
|
-Supramolecule #1: Structure of the Bacillus subtilis hibernating 100S ribosome reve...
| Supramolecule | Name: Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 7.2 |
|---|---|
| Vitrification | Cryogen name: ETHANE |
| Details | Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Average electron dose: 2.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Refinement | Protocol: RIGID BODY FIT |
|---|
