ジャーナル: EMBO J / 年: 2017 タイトル: Structures and dynamics of hibernating ribosomes from mediated by intermolecular interactions of HPF. 著者: Iskander Khusainov / Quentin Vicens / Rustam Ayupov / Konstantin Usachev / Alexander Myasnikov / Angelita Simonetti / Shamil Validov / Bruno Kieffer / Gulnara Yusupova / Marat Yusupov / Yaser Hashem / 要旨: In bacteria, ribosomal hibernation shuts down translation as a response to stress, through reversible binding of stress-induced proteins to ribosomes. This process typically involves the formation of ...In bacteria, ribosomal hibernation shuts down translation as a response to stress, through reversible binding of stress-induced proteins to ribosomes. This process typically involves the formation of 100S ribosome dimers. Here, we present the structures of hibernating ribosomes from human pathogen containing a long variant of the hibernation-promoting factor (SaHPF) that we solved using cryo-electron microscopy. Our reconstructions reveal that the N-terminal domain (NTD) of SaHPF binds to the 30S subunit as observed for shorter variants of HPF in other species. The C-terminal domain (CTD) of SaHPF protrudes out of each ribosome in order to mediate dimerization. Using NMR, we characterized the interactions at the CTD-dimer interface. Secondary interactions are provided by helix 26 of the 16S ribosomal RNA We also show that ribosomes in the 100S particle adopt both rotated and unrotated conformations. Overall, our work illustrates a specific mode of ribosome dimerization by long HPF, a finding that may help improve the selectivity of antimicrobials.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 Staphylococcus aureus (strain NCTC 8325) (黄色ブドウ球菌)
Staphylococcus aureus (strain NCTC 8325) (黄色ブドウ球菌) データ登録者
データ登録者 引用
引用

 構造の表示
構造の表示 ムービービューア
ムービービューア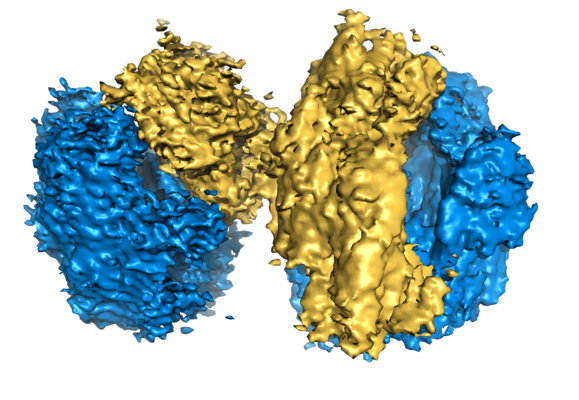
 ダウンロードとリンク
ダウンロードとリンク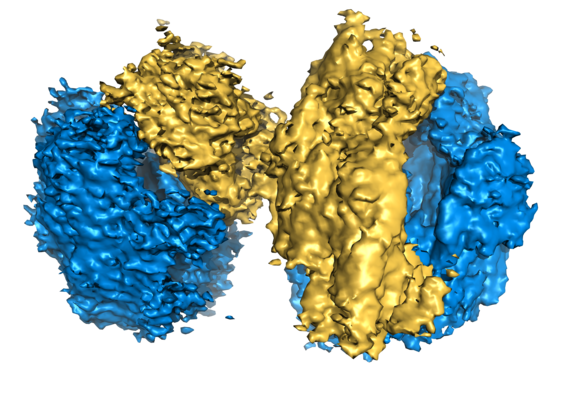 emd_3639.png
emd_3639.png http://ftp.pdbj.org/pub/emdb/structures/EMD-3639
http://ftp.pdbj.org/pub/emdb/structures/EMD-3639












 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN

