ジャーナル: IUCrJ / 年: 2017 タイトル: Single-particle cryo-EM using alignment by classification (ABC): the structure of haemoglobin. 著者: Pavel Afanasyev / Charlotte Seer-Linnemayr / Raimond B G Ravelli / Rishi Matadeen / Sacha De Carlo / Bart Alewijnse / Rodrigo V Portugal / Navraj S Pannu / Michael Schatz / Marin van Heel / 要旨: Single-particle cryogenic electron microscopy (cryo-EM) can now yield near-atomic resolution structures of biological complexes. However, the reference-based alignment algorithms commonly used in ...Single-particle cryogenic electron microscopy (cryo-EM) can now yield near-atomic resolution structures of biological complexes. However, the reference-based alignment algorithms commonly used in cryo-EM suffer from reference bias, limiting their applicability (also known as the 'Einstein from random noise' problem). Low-dose cryo-EM therefore requires robust and objective approaches to reveal the structural information contained in the extremely noisy data, especially when dealing with small structures. A reference-free pipeline is presented for obtaining near-atomic resolution three-dimensional reconstructions from heterogeneous ('four-dimensional') cryo-EM data sets. The methodologies integrated in this pipeline include camera correction, movie-based full-data-set contrast transfer function determination, movie-alignment algorithms, (Fourier-space) multivariate statistical data compression and unsupervised classification, 'random-startup' three-dimensional reconstructions, four-dimensional structural refinements and Fourier shell correlation criteria for evaluating anisotropic resolution. The procedures exclusively use information emerging from the data set itself, without external 'starting models'. Euler-angle assignments are performed by angular reconstitution rather than by the inherently slower projection-matching approaches. The comprehensive 'ABC-4D' pipeline is based on the two-dimensional reference-free 'alignment by classification' (ABC) approach, where similar images in similar orientations are grouped by unsupervised classification. Some fundamental differences between X-ray crystallography single-particle cryo-EM data collection and data processing are discussed. The structure of the giant haemoglobin from at a global resolution of ∼3.8 Å is presented as an example of the use of the ABC-4D procedure.
Reference-free Alignment By Classification (ABC-4D). Camera correction, CTF determination, particle-picking, MSA unsupervised classification, 3D reconstruction, all in IMAGIC-4D.
CTF補正
詳細: Phase flipping of micrograph patches (ctf2d-find)
最終 再構成
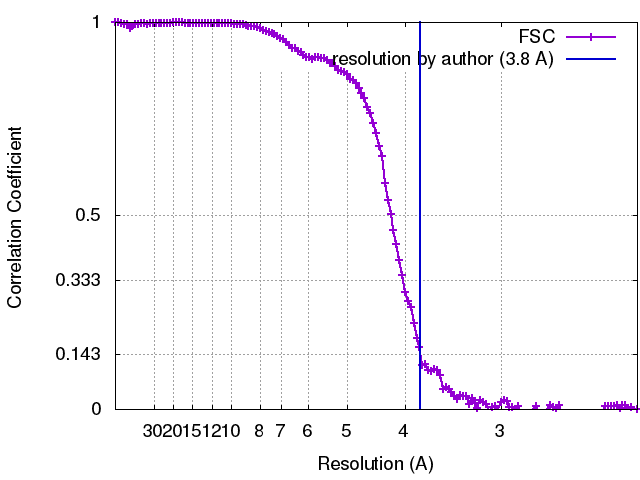
想定した対称性 - 点群: D6 (2回x6回 2面回転対称) アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 3.8 Å / 解像度の算出法: OTHER / ソフトウェア - 名称: Imagic-4D / 使用した粒子像数: 85000
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報 Lumbricus terrestris (無脊椎動物)
Lumbricus terrestris (無脊椎動物) データ登録者
データ登録者 引用
引用




 構造の表示
構造の表示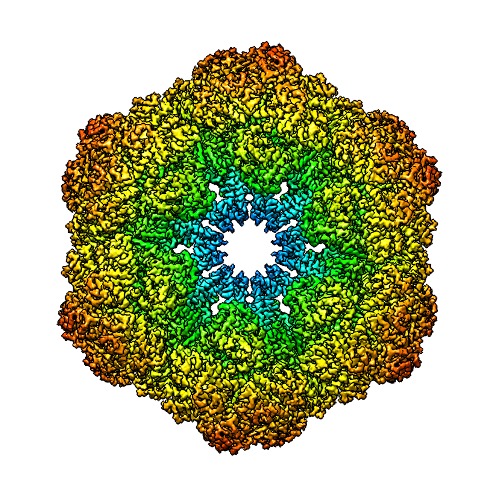
 ダウンロードとリンク
ダウンロードとリンク http://ftp.pdbj.org/pub/emdb/structures/EMD-3434
http://ftp.pdbj.org/pub/emdb/structures/EMD-3434














 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


























 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN