Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-30013 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | V/A-ATPase from Thermus thermophilus | ||||||||||||
 Map data Map data | Cryo-EM map of V/A-ATPase from Thermus thermophilus | ||||||||||||
 Sample Sample |
| ||||||||||||
| Biological species |   Thermus thermophilus HB8 (bacteria) Thermus thermophilus HB8 (bacteria) | ||||||||||||
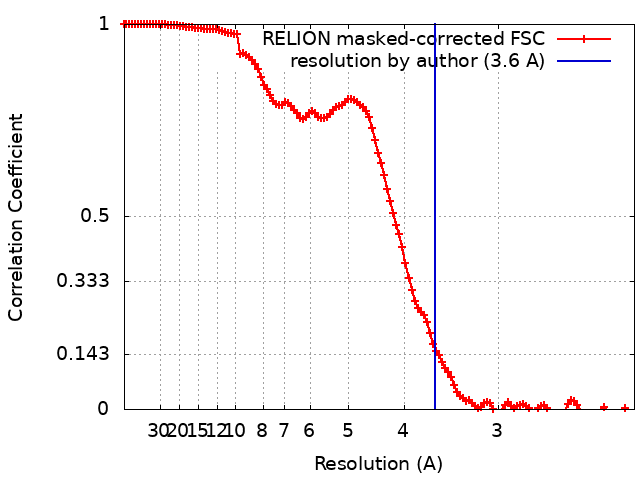
| Method | single particle reconstruction / cryo EM / Resolution: 3.6 Å | ||||||||||||
 Authors Authors | Kishikawa J / Nakanishi A / Furuta A / Kato T / Namba K / Tamakoshi M / Mitsuoka K / Yokoyama K | ||||||||||||
| Funding support |  Japan, 3 items Japan, 3 items
| ||||||||||||
 Citation Citation | Journal: Elife / Year: 2020 Title: Mechanical inhibition of isolated V from V/A-ATPase for proton conductance. Authors: Jun-Ichi Kishikawa / Atsuko Nakanishi / Aya Furuta / Takayuki Kato / Keiichi Namba / Masatada Tamakoshi / Kaoru Mitsuoka / Ken Yokoyama / Abstract: V-ATPase is an energy converting enzyme, coupling ATP hydrolysis/synthesis in the hydrophilic V domain, with proton flow through the V membrane domain, via rotation of the central rotor complex ...V-ATPase is an energy converting enzyme, coupling ATP hydrolysis/synthesis in the hydrophilic V domain, with proton flow through the V membrane domain, via rotation of the central rotor complex relative to the surrounding stator apparatus. Upon dissociation from the V domain, the V domain of the eukaryotic V-ATPase can adopt a physiologically relevant auto-inhibited form in which proton conductance through the V domain is prevented, however the molecular mechanism of this inhibition is not fully understood. Using cryo-electron microscopy, we determined the structure of both the V/A-ATPase and isolated V at near-atomic resolution, respectively. These structures clarify how the isolated V domain adopts the auto-inhibited form and how the complex prevents formation of the inhibited V form. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_30013.map.gz | 12.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-30013-v30.xmlemd-30013.xml | 12.6 KB 12.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_30013_fsc.xml | 12.1 KB | Display | FSC data file |
| Images |  emd_30013.png emd_30013.png | 49.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-30013ftp://ftp.pdbj.org/pub/emdb/structures/EMD-30013 http://ftp.pdbj.org/pub/emdb/structures/EMD-30013ftp://ftp.pdbj.org/pub/emdb/structures/EMD-30013 | HTTPS FTP |
-Related structure data









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_30013.map.gz / Format: CCP4 / Size: 149.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Cryo-EM map of V/A-ATPase from Thermus thermophilus | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : V/A-type ATPase
| Entire | Name: V/A-type ATPase |
|---|---|
| Components |
|
-Supramolecule #1: V/A-type ATPase
| Supramolecule | Name: V/A-type ATPase / type: complex / ID: 1 / Parent: 0 / Details: V/A-type ATPase from Thermus thermophilus |
|---|---|
| Source (natural) | Organism: Thermus thermophilus HB8 (bacteria) |
| Molecular weight | Theoretical: 650 kDa/nm |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3.0 mg/mL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 8 Component:
| |||||||||
| Grid | Model: Quantifoil R1.2/1.3 / Material: MOLYBDENUM / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: AIR | |||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV | |||||||||
| Details | The sample was purified from cell membrane of Thermus thermophilus and incorporated into nanodisc. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON II (4k x 4k) / Detector mode: INTEGRATING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Digitization - Frames/image: 1-34 / Number real images: 3694 / Average exposure time: 2.0 sec. / Average electron dose: 45.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated defocus max: 3.5 µm / Calibrated defocus min: 2.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 75000 |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |