 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-28244: CryoEM characterization of BrxL -- a unique AAA+ phage restrictio... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM characterization of BrxL -- a unique AAA+ phage restriction Factor. | |||||||||
 Map data Map data | combined 1.12 pixel data collected at Hutch with 1.16 pixel data from UW, ie down sampled from 1.12 to 1.16 | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | phage restriction factor / AAA+ protein. BrxL / ANTIMICROBIAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationATP-dependent peptidase activity / protein catabolic process / serine-type endopeptidase activity / proteolysis / ATP binding Similarity search - Function | |||||||||
| Biological species |  Acinetobacter (bacteria) / Acinetobacter sp. NEB 394 (bacteria) Acinetobacter (bacteria) / Acinetobacter sp. NEB 394 (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.6 Å | |||||||||
 Authors Authors | Shen BW / Stoddard BL | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Nucleic Acids Res / Year: 2023 Title: Structure, substrate binding and activity of a unique AAA+ protein: the BrxL phage restriction factor. Authors: Betty W Shen / Lindsey A Doyle / Rachel Werther / Abigail A Westburg / Daniel P Bies / Stephanie I Walter / Yvette A Luyten / Richard D Morgan / Barry L Stoddard / Brett K Kaiser / Abstract: Bacteriophage exclusion ('BREX') systems are multi-protein complexes encoded by a variety of bacteria and archaea that restrict phage by an unknown mechanism. One BREX factor, termed BrxL, has been ...Bacteriophage exclusion ('BREX') systems are multi-protein complexes encoded by a variety of bacteria and archaea that restrict phage by an unknown mechanism. One BREX factor, termed BrxL, has been noted to display sequence similarity to various AAA+ protein factors including Lon protease. In this study we describe multiple CryoEM structures of BrxL that demonstrate it to be a chambered, ATP-dependent DNA binding protein. The largest BrxL assemblage corresponds to a dimer of heptamers in the absence of bound DNA, versus a dimer of hexamers when DNA is bound in its central pore. The protein displays DNA-dependent ATPase activity, and ATP binding promotes assembly of the complex on DNA. Point mutations within several regions of the protein-DNA complex alter one or more in vitro behaviors and activities, including ATPase activity and ATP-dependent association with DNA. However, only the disruption of the ATPase active site fully eliminates phage restriction, indicating that other mutations can still complement BrxL function within the context of an otherwise intact BREX system. BrxL displays significant structural homology to MCM subunits (the replicative helicase in archaea and eukaryotes), implying that it and other BREX factors may collaborate to disrupt initiation of phage DNA replication. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_28244.map.gz | 114.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-28244-v30.xmlemd-28244.xml | 17.7 KB 17.7 KB | Display Display | EMDB header |
| Images | 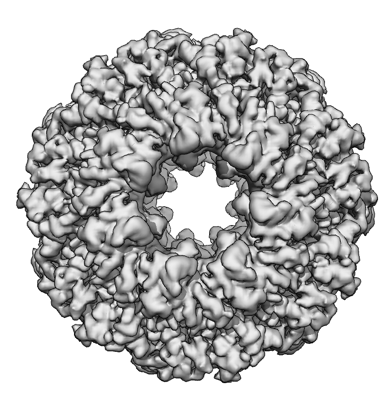 emd_28244.png emd_28244.png | 148.3 KB | ||
| Filedesc metadata | emd-28244.cif.gz | 6.3 KB | ||
| Others | emd_28244_half_map_1.map.gzemd_28244_half_map_2.map.gz | 213.4 MB 213.4 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-28244ftp://ftp.pdbj.org/pub/emdb/structures/EMD-28244 http://ftp.pdbj.org/pub/emdb/structures/EMD-28244ftp://ftp.pdbj.org/pub/emdb/structures/EMD-28244 | HTTPS FTP |
-Related structure data
| Related structure data |  8emcMC  8eilC  8emhC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |

-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_28244.map.gz / Format: CCP4 / Size: 229.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | combined 1.12 pixel data collected at Hutch with 1.16 pixel data from UW, ie down sampled from 1.12 to 1.16 | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.16 Å | ||||||||||||||||||||||||||||||||||||
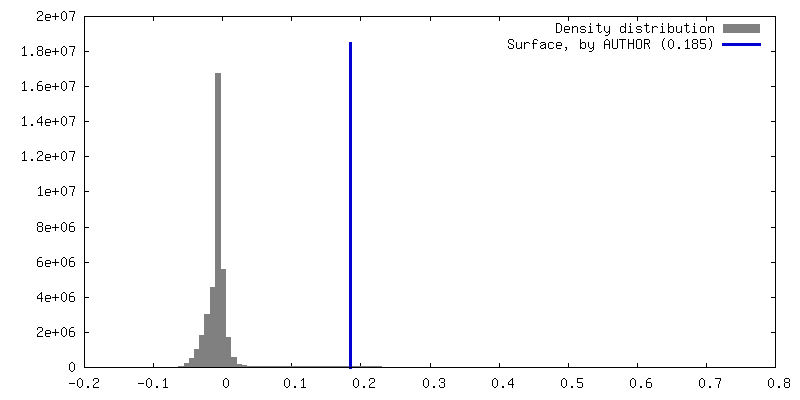
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: half map A
| File | emd_28244_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: half map B
| File | emd_28244_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map B | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





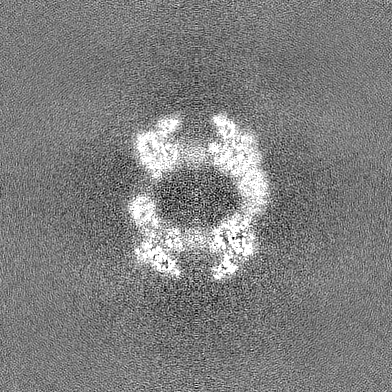

- Sample components
Sample components
-Entire : dimer of heptamer complex of BrxL
| Entire | Name: dimer of heptamer complex of BrxL |
|---|---|
| Components |
|
-Supramolecule #1: dimer of heptamer complex of BrxL
| Supramolecule | Name: dimer of heptamer complex of BrxL / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Acinetobacter (bacteria) |
| Molecular weight | Theoretical: 1.05 MDa |
-Macromolecule #1: Protease Lon-related BREX system protein BrxL
| Macromolecule | Name: Protease Lon-related BREX system protein BrxL / type: protein_or_peptide / ID: 1 / Number of copies: 14 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Acinetobacter sp. NEB 394 (bacteria) |
| Molecular weight | Theoretical: 75.703539 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MESANDKELD QLLNEHFAGR VVRKDLTKLI KEGANVPVYV LEYLLGMYCA SDDPEIIEQG LRNVKTVLAE NYVRPDEAEK VKSLVRERG SYKVIDRVTV KLNERKDKYE ASFSNLGIKD AEISAGIVKE YEKLLVGGIW VIATLSYYFE EGQTSSPFGV S LLKPIQMP ...String: MESANDKELD QLLNEHFAGR VVRKDLTKLI KEGANVPVYV LEYLLGMYCA SDDPEIIEQG LRNVKTVLAE NYVRPDEAEK VKSLVRERG SYKVIDRVTV KLNERKDKYE ASFSNLGIKD AEISAGIVKE YEKLLVGGIW VIATLSYYFE EGQTSSPFGV S LLKPIQMP NMNMDELFSG RAALSTDQWR ESLIRSIGME PASLKEDVQW HLLARMVPFV ENNYNVCELG PRGTGKSHIY KE CSPNSIL VSGGQTTVAN LFYNMSSRRI GLVGLWDVVA FDEVAGISFK DKDGVQIMKD YMASGSFARG REQMEASASM VFV GNINQS VESLVKTSHL LAPFPEAMID SAFFDRFHAY IPGWEIPKMR PEFFTNRYGL IVDYLAEFFR EMRKRSFADS IEKY FKLGN NLNQRDVIAV RKTVSGLMKL LYPHGQFNKE DVRQCLEYAL QVRRRVKEQL KKIGGMEFYD VHFSYIDNDT LEEHF VSVK EQGGGGLIPE GPAKPGFLYT IGLSNKGMPG LYRLELQVTK GSGKLATSGL WNSSSAKEQV KIAFDYFKAN ASRISG GSK VMEHDFHLHV VELQNTGPLS HLALPSLVAF ASGLLGRSVQ SQMVVLGDMS LGGSVTPVES IAECLQVAFD AGAKKVA LP MSSAADIPTI PVELFTKFQT SFYADPVDAV FKGLGVD UniProtKB: Protease Lon-related BREX system protein BrxL |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.6 mg/mL |
|---|---|
| Buffer | pH: 8 / Details: 20 mM TrisHCl, 150 mM NaCl |
| Grid | Model: Quantifoil R1.2/1.3 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 15 sec. / Pretreatment - Atmosphere: AIR |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 298 K / Instrument: FEI VITROBOT MARK IV |
| Details | This sample was mono-dispersed |
- Electron microscopy
Electron microscopy
| Microscope | FEI TALOS ARCTICA |
|---|---|
| Image recording | Film or detector model: DIRECT ELECTRON DE-10 (5k x 4k) / Detector mode: COUNTING / Number grids imaged: 2 / Number real images: 3000 / Average exposure time: 2.0 sec. / Average electron dose: 50.0 e/Å2 Details: 2980 movies at 1.12 pixel was extracted at box size of 406 pix, fourier cropped to box size of 392 pix and combined with 100 movies collected at 1.16 pix size |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 5.0 µm / Nominal defocus min: 1.2 µm / Nominal magnification: 38000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Talos Arctica / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Space: REAL / Protocol: AB INITIO MODEL |
|---|---|
| Output model | PDB-8emc: |
