 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry |  | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
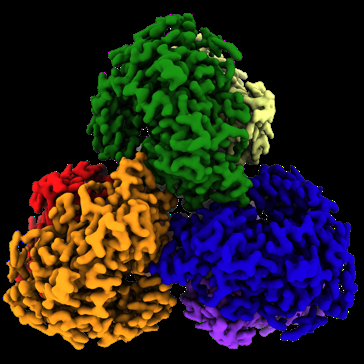
| Title | Structure of the L. blandensis dGTPase in the apo form | ||||||||||||
 Map data Map data | DeepEMhancer post-processed map | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | dGTPase / nucleotide binding / HYDROLASE | ||||||||||||
| Function / homology |  Function and homology information Function and homology information | ||||||||||||
| Biological species |  Leeuwenhoekiella blandensis MED217 (bacteria) Leeuwenhoekiella blandensis MED217 (bacteria) | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.1 Å | ||||||||||||
 Authors Authors | Klemm BP / Sikkema AP | ||||||||||||
| Funding support |  United States, 3 items United States, 3 items
| ||||||||||||
 Citation Citation | Journal: J Biol Chem / Year: 2022 Title: High-resolution structures of the SAMHD1 dGTPase homolog from Leeuwenhoekiella blandensis reveal a novel mechanism of allosteric activation by dATP. Authors: Bradley P Klemm / Andrew P Sikkema / Allen L Hsu / James C Horng / Traci M Tanaka Hall / Mario J Borgnia / Roel M Schaaper / Abstract: Deoxynucleoside triphosphate (dNTP) triphosphohydrolases (dNTPases) are important enzymes that may perform multiple functions in the cell, including regulating the dNTP pools and contributing to ...Deoxynucleoside triphosphate (dNTP) triphosphohydrolases (dNTPases) are important enzymes that may perform multiple functions in the cell, including regulating the dNTP pools and contributing to innate immunity against viruses. Among the homologs that are best studied are human sterile alpha motif and HD domain-containing protein 1 (SAMHD1), a tetrameric dNTPase, and the hexameric Escherichia coli dGTPase; however, it is unclear whether these are representative of all dNTPases given their wide distribution throughout life. Here, we investigated a hexameric homolog from the marine bacterium Leeuwenhoekiella blandensis, revealing that it is a dGTPase that is subject to allosteric activation by dATP, specifically. Allosteric regulation mediated solely by dATP represents a novel regulatory feature among dNTPases that may facilitate maintenance of cellular dNTP pools in L. blandensis. We present high-resolution X-ray crystallographic structures (1.80-2.26 Å) in catalytically important conformations as well as cryo-EM structures (2.1-2.7 Å) of the enzyme bound to dGTP and dATP ligands. The structures, the highest resolution cryo-EM structures of any SAMHD1-like dNTPase to date, reveal an intact metal-binding site with the dGTP substrate coordinated to three metal ions. These structural and biochemical data yield insights into the catalytic mechanism and support a conserved catalytic mechanism for the tetrameric and hexameric dNTPase homologs. We conclude that the allosteric activation by dATP appears to rely on structural connectivity between the allosteric and active sites, as opposed to the changes in oligomeric state upon ligand binding used by SAMHD1. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_26126.map.gz | 159.1 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-26126-v30.xmlemd-26126.xml | 27.3 KB 27.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_26126_fsc.xml | 13 KB | Display | FSC data file |
| Images |  emd_26126.png emd_26126.png | 135.4 KB | ||
| Masks | emd_26126_msk_1.map | 190.1 MB | Mask map | |
| Filedesc metadata | emd-26126.cif.gz | 6.9 KB | ||
| Others | emd_26126_additional_1.map.gzemd_26126_additional_2.map.gzemd_26126_additional_3.map.gzemd_26126_half_map_1.map.gzemd_26126_half_map_2.map.gz | 149.4 MB 19.6 MB 176.2 MB 150.8 MB 150.8 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-26126ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26126 http://ftp.pdbj.org/pub/emdb/structures/EMD-26126ftp://ftp.pdbj.org/pub/emdb/structures/EMD-26126 | HTTPS FTP |
-Related structure data
| Related structure data |  7tu5MC  7tu0C  7tu1C  7tu2C  7tu3C  7tu4C  7tu6C  7tu7C  7tu8C C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |



-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_26126.map.gz / Format: CCP4 / Size: 190.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | DeepEMhancer post-processed map | ||||||||||||||||||||||||||||||||||||
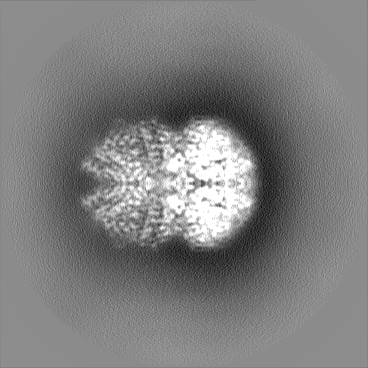
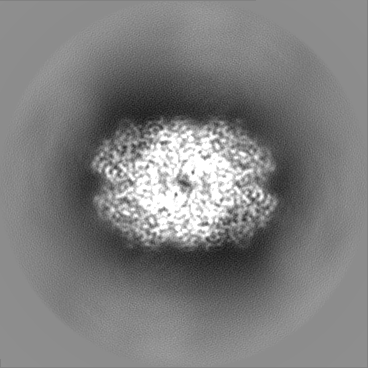
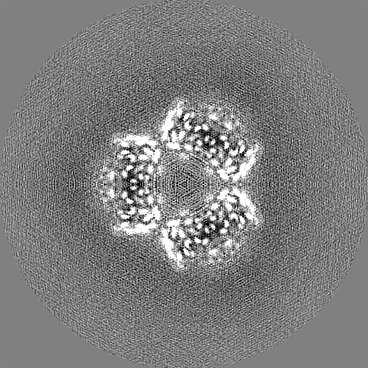
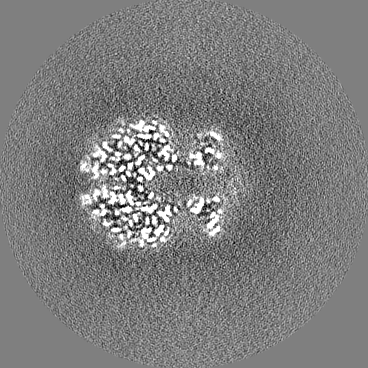
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.646 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_26126_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
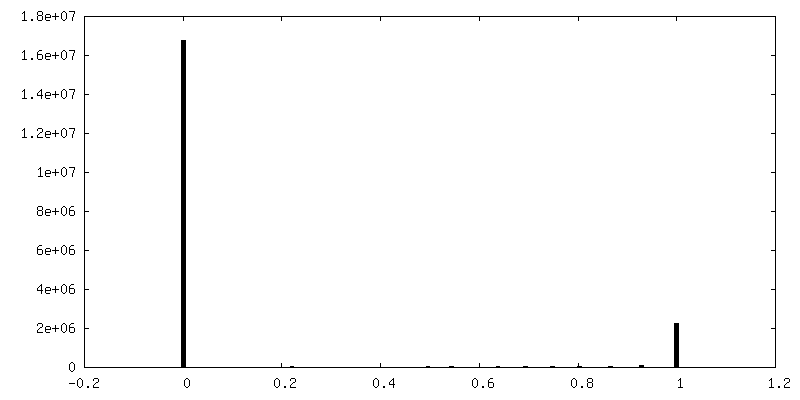
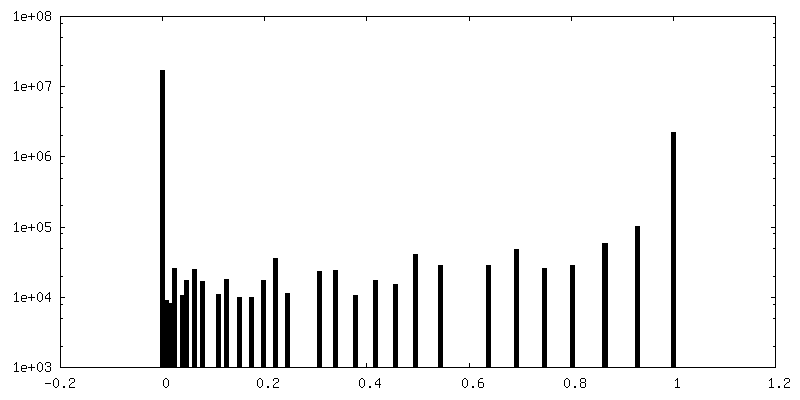
| Density Histograms |






-Additional map: Full-map from RELION refinement
| File | emd_26126_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Full-map from RELION refinement | ||||||||||||
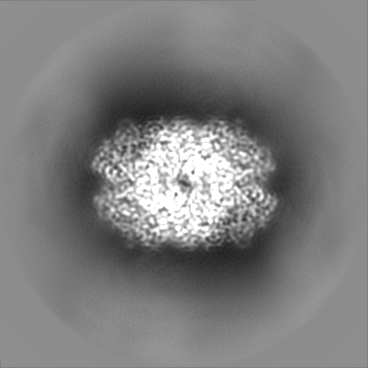
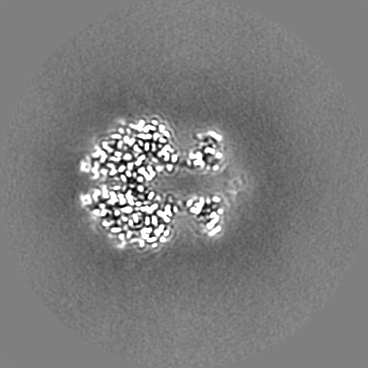
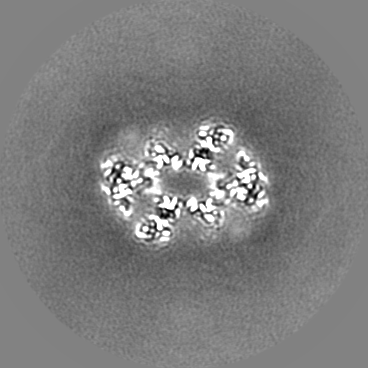
| Projections & Slices |
| ||||||||||||
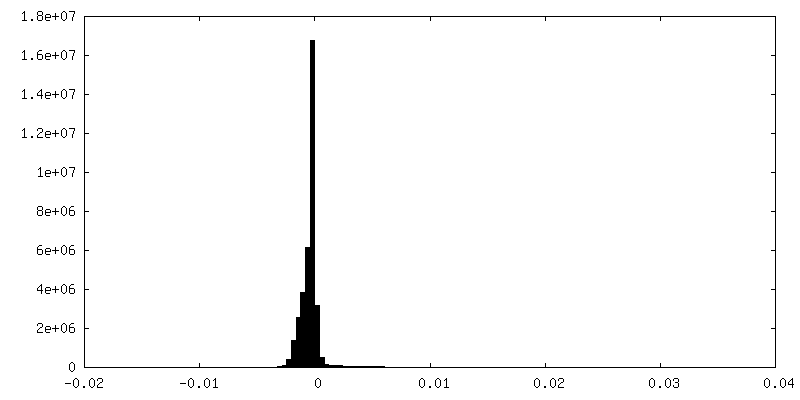
| Density Histograms |



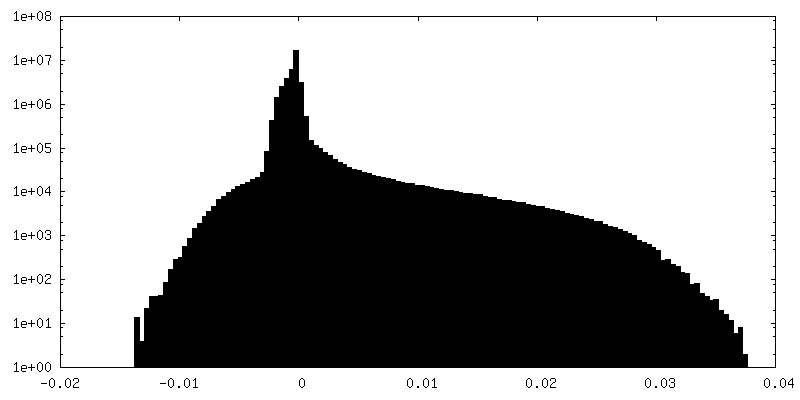
-Additional map: RELION post-processed map
| File | emd_26126_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | RELION post-processed map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |





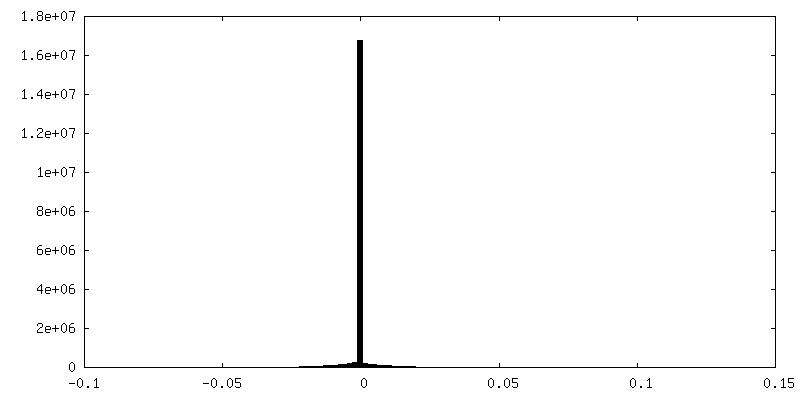
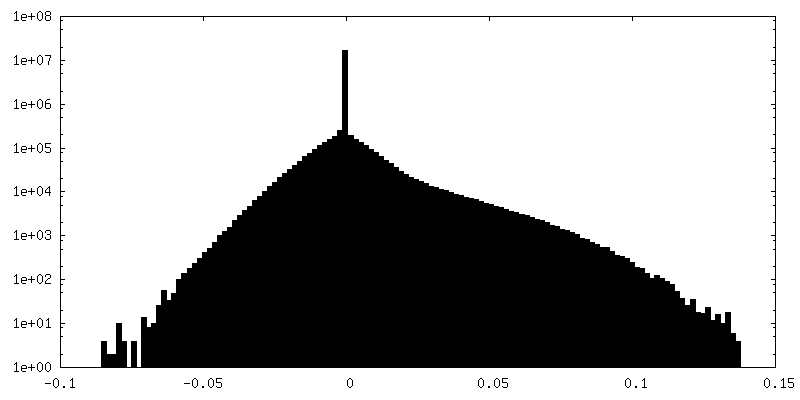
-Additional map: PHENIX auto-sharpened map
| File | emd_26126_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | PHENIX auto-sharpened map | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: Half-map 1
| File | emd_26126_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

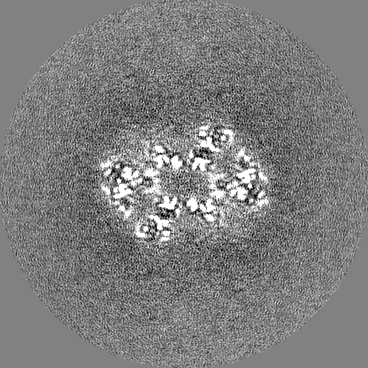
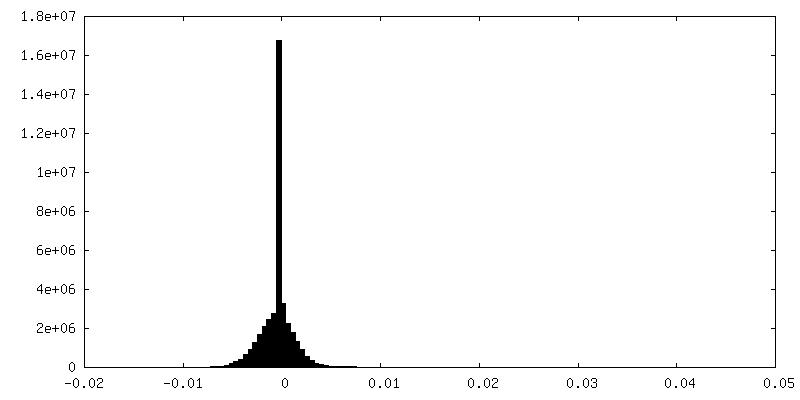
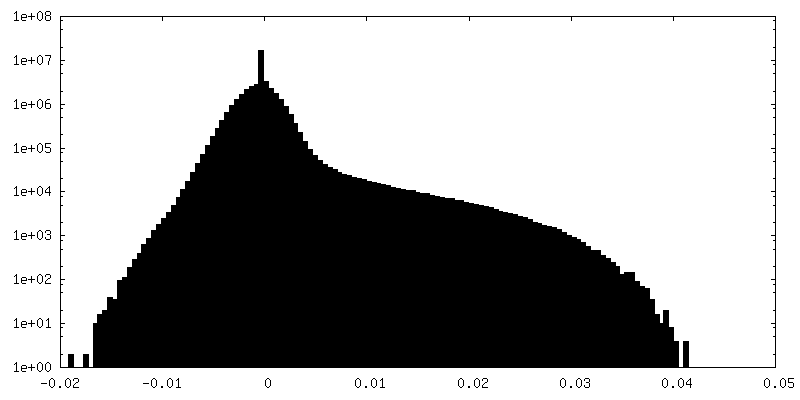
-Half map: Half-map 2
| File | emd_26126_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
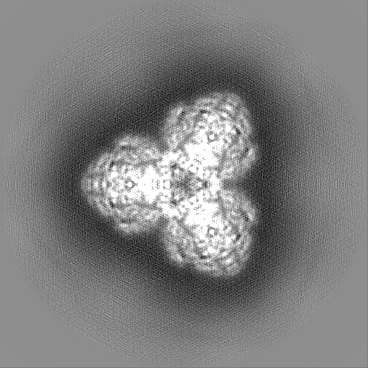
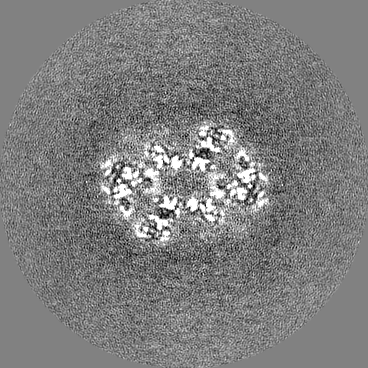
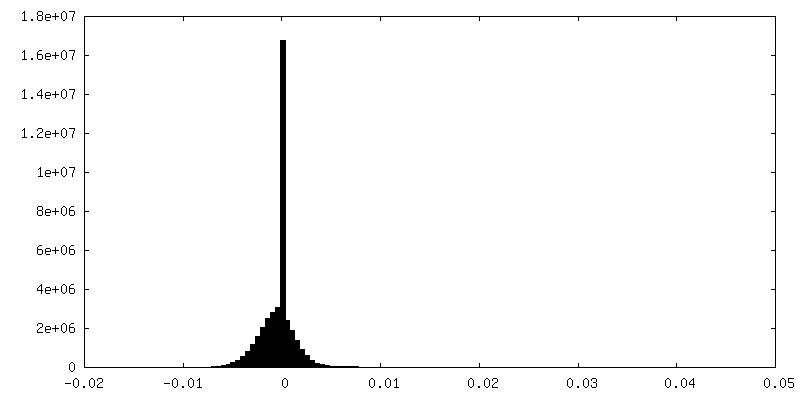
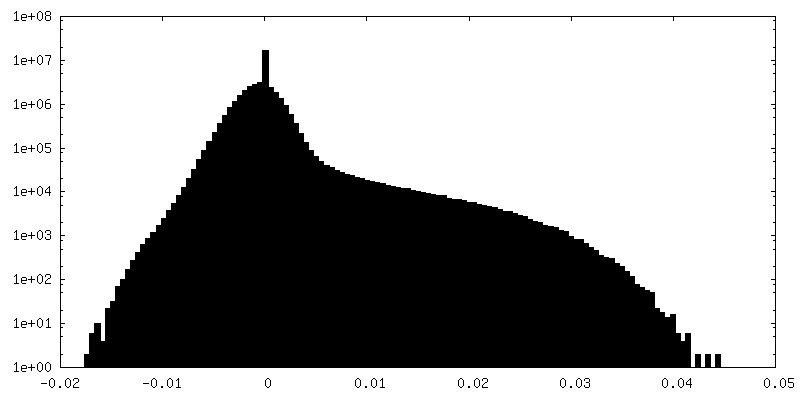
- Sample components
Sample components
-Entire : dGTP triphosphohydrolase
| Entire | Name: dGTP triphosphohydrolase |
|---|---|
| Components |
|
-Supramolecule #1: dGTP triphosphohydrolase
| Supramolecule | Name: dGTP triphosphohydrolase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Leeuwenhoekiella blandensis MED217 (bacteria) |
-Macromolecule #1: dGTP triphosphohydrolase
| Macromolecule | Name: dGTP triphosphohydrolase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO / EC number: dGTPase |
|---|---|
| Source (natural) | Organism: Leeuwenhoekiella blandensis MED217 (bacteria) / Strain: CECT 7118 / CCUG 51940 / MED217 |
| Molecular weight | Theoretical: 52.723242 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MHHHHHHSSG VDLGTENLYF QSNANWEHLL SLKRQGDTAK RLRIEQDDTR LGFEVDYDRI IFSAPFRSLQ DKTQVIPLSK TDFVHTRLT HSLEVSVVGR SLGRMVGKKL LEKYPHLEQV YGYKFNDFGA IVAAAALAHD IGNPPFGHSG EKAIGEFFKN G YGKRYKDS ...String: MHHHHHHSSG VDLGTENLYF QSNANWEHLL SLKRQGDTAK RLRIEQDDTR LGFEVDYDRI IFSAPFRSLQ DKTQVIPLSK TDFVHTRLT HSLEVSVVGR SLGRMVGKKL LEKYPHLEQV YGYKFNDFGA IVAAAALAHD IGNPPFGHSG EKAIGEFFKN G YGKRYKDS LTAKEYQDLI KFEGNANGFK VLSQSKPGAQ GGLRLSYATL GAFMKYPKES LPHKPSDHIA DKKYGFFQSE RA LFEDVAQ ELGLLKRSTT DDVSWSRHPL AYLVEAADDI CYTIIDFEDG INLGLIPEEY ALEYMVKLVG QTIDRNKYNA LQE TSDRVS YLRALAIGTL INESVDTFMK YEEEILAGTF DQSLIDKSNY QAQITDIINL SIERIYNSRE VIEKEIAGYE ILST LLEAR CRALDNNDTH YNQLIQQLLA PNDHSEKSLY ENLIQICAEV STMTDGKALR NYKKIKGLD UniProtKB: dGTP triphosphohydrolase |
-Macromolecule #2: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 2 / Number of copies: 6 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 6 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE | ||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 290 K / Instrument: LEICA EM GP / Details: 3.5 second blot time (front). | ||||||||||||
| Details | Hexamer concentration is listed. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Details | Defocus values as determined by CTFFIND-4.1 |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Number grids imaged: 1 / Number real images: 3780 / Average exposure time: 4.0 sec. / Average electron dose: 58.6 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.5 µm / Nominal magnification: 215000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
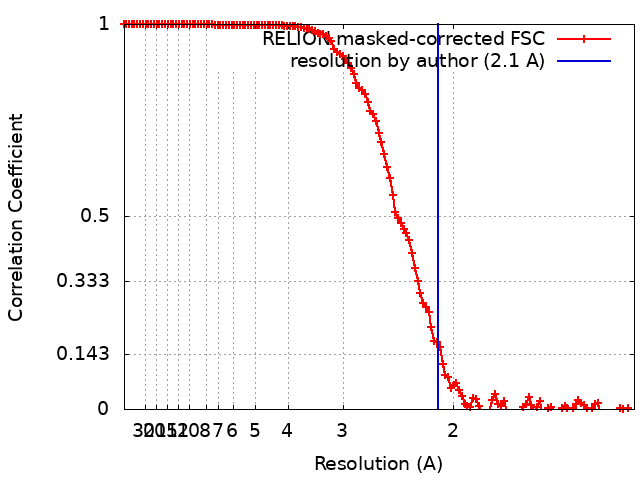
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | The hexameric biological assembly of PDB ID: 3BG2 was used as the initial input model to fit into the EM map. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
| Output model | PDB-7tu5: |

