 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-25791 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | cryo-EM structure of MBP-KIX-apoferritin | ||||||||||||
 Map data Map data | cryo-EM structure of MBP-KIX-apoferritin | ||||||||||||
 Sample Sample |
| ||||||||||||
 Keywords Keywords | Protein Engineering / Simulation / Peptide Therapeutics / Acute Myeloid Leukemia / STRUCTURAL PROTEIN | ||||||||||||
| Function / homology |  Function and homology information Function and homology informationIron uptake and transport / Golgi Associated Vesicle Biogenesis / N-terminal peptidyl-lysine acetylation / peptide lactyltransferase (CoA-dependent) activity / Phosphorylation of CLOCK, acetylation of BMAL1 (ARNTL) at target gene promoters / NFE2L2 regulating ER-stress associated genes / NFE2L2 regulating inflammation associated genes / Activation of the TFAP2 (AP-2) family of transcription factors / The CRY:PER:kinase complex represses transactivation by the BMAL:CLOCK (ARNTL:CLOCK) complex / histone H3K18 acetyltransferase activity ...Iron uptake and transport / Golgi Associated Vesicle Biogenesis / N-terminal peptidyl-lysine acetylation / peptide lactyltransferase (CoA-dependent) activity / Phosphorylation of CLOCK, acetylation of BMAL1 (ARNTL) at target gene promoters / NFE2L2 regulating ER-stress associated genes / NFE2L2 regulating inflammation associated genes / Activation of the TFAP2 (AP-2) family of transcription factors / The CRY:PER:kinase complex represses transactivation by the BMAL:CLOCK (ARNTL:CLOCK) complex / histone H3K18 acetyltransferase activity / histone H3K27 acetyltransferase activity / LRR FLII-interacting protein 1 (LRRFIP1) activates type I IFN production / NFE2L2 regulates pentose phosphate pathway genes / regulation of smoothened signaling pathway / NFE2L2 regulating MDR associated enzymes / homeostatic process / MRF binding / negative regulation of transcription by RNA polymerase I / Regulation of FOXO transcriptional activity by acetylation / embryonic digit morphogenesis / Regulation of gene expression in late stage (branching morphogenesis) pancreatic bud precursor cells / RUNX3 regulates NOTCH signaling / NOTCH4 Intracellular Domain Regulates Transcription / RUNX1 regulates transcription of genes involved in differentiation of myeloid cells / Regulation of NFE2L2 gene expression / Regulation of gene expression by Hypoxia-inducible Factor / Nuclear events mediated by NFE2L2 / Phosphorylated BMAL1:CLOCK (ARNTL:CLOCK) activates expression of core clock genes / TRAF6 mediated IRF7 activation / NOTCH3 Intracellular Domain Regulates Transcription / NFE2L2 regulating tumorigenic genes / NFE2L2 regulating anti-oxidant/detoxification enzymes / protein acetylation / Notch-HLH transcription pathway / Formation of paraxial mesoderm / acetyltransferase activity / stimulatory C-type lectin receptor signaling pathway / histone acetyltransferase activity / FOXO-mediated transcription of cell death genes / positive regulation of transforming growth factor beta receptor signaling pathway / Zygotic genome activation (ZGA) / ferroxidase / TP53 Regulates Transcription of Genes Involved in Cytochrome C Release / autolysosome / negative regulation of ferroptosis / ferroxidase activity / histone acetyltransferase complex / negative regulation of fibroblast proliferation / Attenuation phase / protein-lysine-acetyltransferase activity / cAMP/PKA signal transduction / histone acetyltransferase / positive regulation of double-strand break repair via homologous recombination / canonical NF-kappaB signal transduction / regulation of cellular response to heat / Regulation of lipid metabolism by PPARalpha / NPAS4 regulates expression of target genes / endocytic vesicle lumen / Neutrophil degranulation / BMAL1:CLOCK,NPAS2 activates circadian expression / Transcriptional and post-translational regulation of MITF-M expression and activity / RORA,B,C and NR1D1 (REV-ERBA) regulate gene expression / Activation of gene expression by SREBF (SREBP) / SUMOylation of transcription cofactors / Expression of BMAL (ARNTL), CLOCK, and NPAS2 / Transferases; Acyltransferases; Transferring groups other than aminoacyl groups / ferric iron binding / CD209 (DC-SIGN) signaling / cellular response to nutrient levels / autophagosome / Heme signaling / iron ion transport / PPARA activates gene expression / Cytoprotection by HMOX1 / Transcriptional activation of mitochondrial biogenesis / protein destabilization / Formation of the beta-catenin:TCF transactivating complex / positive regulation of protein localization to nucleus / ferrous iron binding / Transcriptional regulation of white adipocyte differentiation / chromatin DNA binding / Evasion by RSV of host interferon responses / NOTCH1 Intracellular Domain Regulates Transcription / Pre-NOTCH Transcription and Translation / tau protein binding / Activation of anterior HOX genes in hindbrain development during early embryogenesis / transcription coactivator binding / Constitutive Signaling by NOTCH1 PEST Domain Mutants / Constitutive Signaling by NOTCH1 HD+PEST Domain Mutants / cellular response to UV / p53 binding / transcription corepressor activity / rhythmic process / HATs acetylate histones / MLL4 and MLL3 complexes regulate expression of PPARG target genes in adipogenesis and hepatic steatosis / protein-containing complex assembly / TRAF3-dependent IRF activation pathway / transcription regulator complex / Estrogen-dependent gene expression / DNA-binding transcription factor binding Similarity search - Function | ||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.57 Å | ||||||||||||
 Authors Authors | Zhang K / Horikoshi N | ||||||||||||
| Funding support |  United States, 3 items United States, 3 items
| ||||||||||||
 Citation Citation | Journal: ACS Cent Sci / Year: 2022 Title: Cryo-EM, Protein Engineering, and Simulation Enable the Development of Peptide Therapeutics against Acute Myeloid Leukemia. Authors: Kaiming Zhang / Naoki Horikoshi / Shanshan Li / Alexander S Powers / Mikhail A Hameedi / Grigore D Pintilie / Hee-Don Chae / Yousuf A Khan / Carl-Mikael Suomivuori / Ron O Dror / Kathleen M ...Authors: Kaiming Zhang / Naoki Horikoshi / Shanshan Li / Alexander S Powers / Mikhail A Hameedi / Grigore D Pintilie / Hee-Don Chae / Yousuf A Khan / Carl-Mikael Suomivuori / Ron O Dror / Kathleen M Sakamoto / Wah Chiu / Soichi Wakatsuki /   Abstract: Cryogenic electron microscopy (cryo-EM) has emerged as a viable structural tool for molecular therapeutics development against human diseases. However, it remains a challenge to determine structures ...Cryogenic electron microscopy (cryo-EM) has emerged as a viable structural tool for molecular therapeutics development against human diseases. However, it remains a challenge to determine structures of proteins that are flexible and smaller than 30 kDa. The 11 kDa KIX domain of CREB-binding protein (CBP), a potential therapeutic target for acute myeloid leukemia and other cancers, is a protein which has defied structure-based inhibitor design. Here, we develop an experimental approach to overcome the size limitation by engineering a protein double-shell to sandwich the KIX domain between apoferritin as the inner shell and maltose-binding protein as the outer shell. To assist homogeneous orientations of the target, disulfide bonds are introduced at the target-apoferritin interface, resulting in a cryo-EM structure at 2.6 Å resolution. We used molecular dynamics simulations to design peptides that block the interaction of the KIX domain of CBP with the intrinsically disordered pKID domain of CREB. The double-shell design allows for fluorescence polarization assays confirming the binding between the KIX domain in the double-shell and these interacting peptides. Further cryo-EM analysis reveals a helix-helix interaction between a single KIX helix and the best peptide, providing a possible strategy for developments of next-generation inhibitors. | ||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_25791.map.gz | 108.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-25791-v30.xmlemd-25791.xml | 12.7 KB 12.7 KB | Display Display | EMDB header |
| Images | 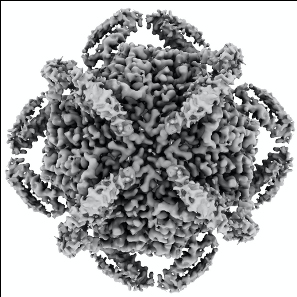 emd_25791.png emd_25791.png | 101 KB | ||
| Filedesc metadata | emd-25791.cif.gz | 5.7 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25791ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25791 http://ftp.pdbj.org/pub/emdb/structures/EMD-25791ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25791 | HTTPS FTP |
-Related structure data
| Related structure data |  7tb3MC  7tbhC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


























-Map
| File | Download / File: emd_25791.map.gz / Format: CCP4 / Size: 216 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | cryo-EM structure of MBP-KIX-apoferritin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.82 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : MBP-KIX-apoferritin
| Entire | Name: MBP-KIX-apoferritin |
|---|---|
| Components |
|
-Supramolecule #1: MBP-KIX-apoferritin
| Supramolecule | Name: MBP-KIX-apoferritin / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 1.7 MDa |
-Macromolecule #1: Isoform 2 of CREB-binding protein,Ferritin heavy chain, N-termina...
| Macromolecule | Name: Isoform 2 of CREB-binding protein,Ferritin heavy chain, N-terminally processed type: protein_or_peptide / ID: 1 / Number of copies: 24 / Enantiomer: LEVO / EC number: histone acetyltransferase |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 30.408438 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: HMGVRKGWHE HVTQDLRSHL VHKLVQAIFP CPDPCALKDR RMENLVAYAK KVEGDMYESA NSRDEYYHLL AEKIYKIQKE LEEKSQVRQ NYHQDAEAAI NRQINLELYA SYVYLSMSCY FDRDDVALKN FAKYFLHQSH EEREHAEKLM KLQNQRGGRI F LQDIKKPD ...String: HMGVRKGWHE HVTQDLRSHL VHKLVQAIFP CPDPCALKDR RMENLVAYAK KVEGDMYESA NSRDEYYHLL AEKIYKIQKE LEEKSQVRQ NYHQDAEAAI NRQINLELYA SYVYLSMSCY FDRDDVALKN FAKYFLHQSH EEREHAEKLM KLQNQRGGRI F LQDIKKPD RDDWCSGLNA MESALHLEKS VNQSLLELHK LATDKNDPHL CDFIETYYLS EQVKSIKELG DHVTNLRKMG AP CAGMAEY LFDKHTLGHG DES UniProtKB: CREB-binding protein, Ferritin heavy chain |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 4 mg/mL |
|---|---|
| Buffer | pH: 8 |
| Grid | Model: Quantifoil R2/1 / Support film - Material: CARBON / Support film - topology: HOLEY |
| Vitrification | Cryogen name: ETHANE |
| Details | MBP-KIX-apoferritin |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 1915 / Average exposure time: 6.0 sec. / Average electron dose: 43.8 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 0.3 µm / Nominal magnification: 165000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Refinement | Protocol: OTHER / Target criteria: Q-score |
|---|---|
| Output model | PDB-7tb3: |
