 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-25582 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CryoEM structure of the crosslinked Rix7 AAA-ATPase | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | CryoEM / AAA-ATPase / ribosome biogenesis / substrate translocation / ribosomal protein | |||||||||
| Function / homology |  Function and homology information Function and homology informationpreribosome binding / peroxisome organization / ribosome biogenesis / ATP hydrolysis activity / RNA binding / ATP binding / nucleus Similarity search - Function | |||||||||
| Biological species |  Chaetomium thermophilum (fungus) / synthetic construct (others) Chaetomium thermophilum (fungus) / synthetic construct (others) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.67 Å | |||||||||
 Authors Authors | Kocaman S / Stanley RE | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: PNAS Nexus / Year: 2022 Title: Communication network within the essential AAA-ATPase Rix7 drives ribosome assembly. Authors: Seda Kocaman / Yu-Hua Lo / Juno M Krahn / Mack Sobhany / Venkata P Dandey / Matthew L Petrovich / Suhas K Etigunta / Jason G Williams / Leesa J Deterding / Mario J Borgnia / Robin E Stanley / Abstract: Rix7 is an essential AAA+ ATPase that functions during the early stages of ribosome biogenesis. Rix7 is composed of three domains including an N-terminal domain (NTD) and two AAA+ domains (D1 and ...Rix7 is an essential AAA+ ATPase that functions during the early stages of ribosome biogenesis. Rix7 is composed of three domains including an N-terminal domain (NTD) and two AAA+ domains (D1 and D2) that assemble into an asymmetric stacked hexamer. It was recently established that Rix7 is a presumed protein translocase that removes substrates from preribosomes by translocating them through its central pore. However, how the different domains of Rix7 coordinate their activities within the overall hexameric structure was unknown. We captured cryo-electron microscopy (EM) structures of single and double Walker B variants of full length Rix7. The disordered NTD was not visible in the cryo-EM reconstructions, but cross-linking mass spectrometry revealed that the NTD can associate with the central channel in vitro. Deletion of the disordered NTD enabled us to obtain a structure of the Rix7 hexamer to 2.9 Å resolution, providing high resolution details of critical motifs involved in substrate translocation and interdomain communication. This structure coupled with cell-based assays established that the linker connecting the D1 and D2 domains as well as the pore loops lining the central channel are essential for formation of the large ribosomal subunit. Together, our work shows that Rix7 utilizes a complex communication network to drive ribosome biogenesis. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_25582.map.gz | 8 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-25582-v30.xmlemd-25582.xml | 11.6 KB 11.6 KB | Display Display | EMDB header |
| Images | 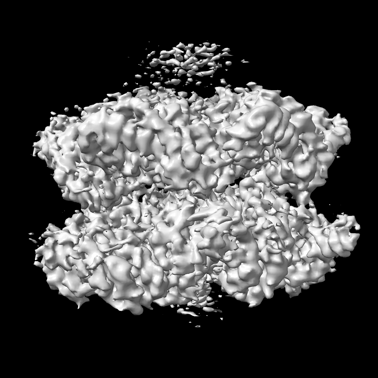 emd_25582.png emd_25582.png | 94.4 KB | ||
| Filedesc metadata | emd-25582.cif.gz | 5.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25582ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25582 http://ftp.pdbj.org/pub/emdb/structures/EMD-25582ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25582 | HTTPS FTP |
-Related structure data
| Related structure data |  7t0vMC  7swlC  7t3iC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |



-Map
| File | Download / File: emd_25582.map.gz / Format: CCP4 / Size: 9.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.03509 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















-Supplemental data
- Sample components
Sample components
-Entire : Cryoem structure of the crosslinked Rix7 AAA-ATPase
| Entire | Name: Cryoem structure of the crosslinked Rix7 AAA-ATPase |
|---|---|
| Components |
|
-Supramolecule #1: Cryoem structure of the crosslinked Rix7 AAA-ATPase
| Supramolecule | Name: Cryoem structure of the crosslinked Rix7 AAA-ATPase / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: Chaetomium thermophilum (fungus) |
| Molecular weight | Theoretical: 100 KDa |
-Macromolecule #1: Rix7
| Macromolecule | Name: Rix7 / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Chaetomium thermophilum (fungus) |
| Molecular weight | Theoretical: 89.418266 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MSRRPTLRLG LDRDVYNIVL NLEQQGTDEN GKRPRLTVDY VYDTIKRSNS SLARQKKRML EDSIERVLAV RKEQAKAEEE TDSDDLIEA QERERERQKA AQAQRDANLL NRQIAKSWGF ASSPGAKAAD GEKGTDTGSI ATPAPATPAV AENMAADTPT T STGPVLPA ...String: MSRRPTLRLG LDRDVYNIVL NLEQQGTDEN GKRPRLTVDY VYDTIKRSNS SLARQKKRML EDSIERVLAV RKEQAKAEEE TDSDDLIEA QERERERQKA AQAQRDANLL NRQIAKSWGF ASSPGAKAAD GEKGTDTGSI ATPAPATPAV AENMAADTPT T STGPVLPA SSTDRQPNGE PRPKKRKAAP KEIDRTPPTK VSILDIAGVD DTLQRLLKEV WFPLRGGEAC EKMGYRYDNG VL LHGPSGC GKTTLAHAIA GSIGVAFIPV SAPSVIGGTS GESEKNIRDV FDEAIRLAPC LIFLDQIDAI AGRRESANKG MES RIVAEI MNGMDRIRQN TPLGKNVVVL AATNRPEFLD PAIRRRFSVE IDMGMPSERA REQILRSLTR DLSLADDINF KELA KMTPG YVGSDLQYVV KAAVSESFQA NIDSLLAQAR AKHPADHLAN VSQPQRDWLL LEAHRDEEVS WPSTKITMEQ FRKAV SLVQ PASKREGFST IPDTTWSHVG ALEDVRKKLE MSIIGPIKNP ELFTRVGIKP AAGILLWGPP GCGKTLVAKA VANESK ANF ISIKGPELLN KYVGESERAV RQLFSRAKSS APCILFFDQM DALVPRRDDS LSDASARVVN TLLTELDGVG DRSGIYV IG ATNRPDMIDE AIRRPGRLGT SIYVGLPSAE DRVKILKTLY RNTVKAPKKR EGTNGEDVDM TDAAAEQQHQ GTTDADLE K VALDLRCTGF SGADLGNLMQ AAAQACLERV YTQRQQKRKE GGSVAEEEEI EPVITMEDWE KALNEVKPSV KDPEKYMHS GFAAALEHHH HHH UniProtKB: Peroxisomal ATPase PEX1 |
-Macromolecule #2: polyvaline
| Macromolecule | Name: polyvaline / type: protein_or_peptide / ID: 2 / Number of copies: 1 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Theoretical: 2.298024 KDa |
| Sequence | String: VVVVVVVVVV VVVVVVVVVV VVV |
-Macromolecule #3: ADENOSINE-5'-TRIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-TRIPHOSPHATE / type: ligand / ID: 3 / Number of copies: 9 / Formula: ATP |
|---|---|
| Molecular weight | Theoretical: 507.181 Da |
| Chemical component information |  ChemComp-ATP: |
-Macromolecule #4: MAGNESIUM ION
| Macromolecule | Name: MAGNESIUM ION / type: ligand / ID: 4 / Number of copies: 9 / Formula: MG |
|---|---|
| Molecular weight | Theoretical: 24.305 Da |
-Macromolecule #5: ADENOSINE-5'-DIPHOSPHATE
| Macromolecule | Name: ADENOSINE-5'-DIPHOSPHATE / type: ligand / ID: 5 / Number of copies: 2 / Formula: ADP |
|---|---|
| Molecular weight | Theoretical: 427.201 Da |
| Chemical component information |  ChemComp-ADP: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 |
|---|---|
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Average electron dose: 60.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
-Image processing
| Startup model | Type of model: PDB ENTRY |
|---|---|
| Final reconstruction | Resolution.type: BY AUTHOR / Resolution: 3.67 Å / Resolution method: FSC 0.143 CUT-OFF / Number images used: 201000 |
| Initial angle assignment | Type: MAXIMUM LIKELIHOOD |
| Final angle assignment | Type: MAXIMUM LIKELIHOOD |
