ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-25481 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | 2.6 A structure of the nucleosome from a class without Chd1 | |||||||||
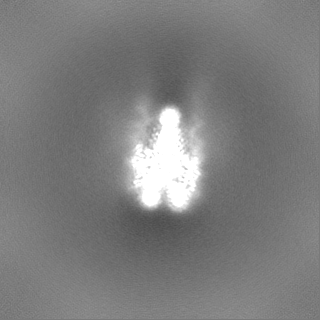
 マップデータ マップデータ | Sharpened 2.6A map in an orientation aligned to a common reference. | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | CHD1 / chromatin remodeling / ATPase / DBD / nucleosome / remodeling / transcription / DNA BINDING PROTEIN | |||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報structural constituent of chromatin / nucleosome / heterochromatin formation / nucleosome assembly / protein heterodimerization activity / DNA binding / nucleus 類似検索 - 分子機能 | |||||||||
| 生物種 |  | |||||||||
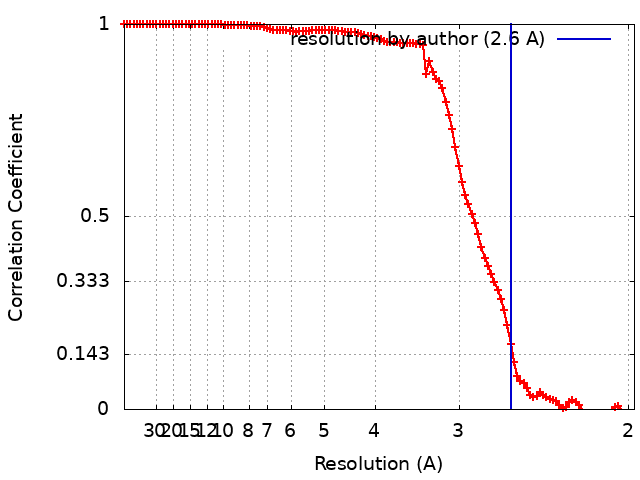
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 2.6 Å | |||||||||
 データ登録者 データ登録者 | Nodelman IM / Das S / Faustino AM / Fried SD / Bowman GD / Armache J-P | |||||||||
| 資金援助 |  米国, 1件 米国, 1件
| |||||||||
 引用 引用 | ジャーナル: Nat Struct Mol Biol / 年: 2022 タイトル: Nucleosome recognition and DNA distortion by the Chd1 remodeler in a nucleotide-free state. 著者: Ilana M Nodelman / Sayan Das / Anneliese M Faustino / Stephen D Fried / Gregory D Bowman / Jean-Paul Armache / 要旨: Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free ...Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free state, determined by cryo-EM to 2.3 Å resolution. The remodeler stimulates the nucleosome to absorb an additional nucleotide on each strand at two different locations: on the tracking strand within the ATPase binding site and on the guide strand one helical turn from the ATPase motor. Remarkably, the additional nucleotide on the tracking strand is associated with a local transformation toward an A-form geometry, explaining how sequential ratcheting of each DNA strand occurs. The structure also reveals a histone-binding motif, ChEx, which can block opposing remodelers on the nucleosome and may allow Chd1 to participate in histone reorganization during transcription. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |
- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_25481.map.gz | 62 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-25481-v30.xmlemd-25481.xml | 26.4 KB 26.4 KB | 表示 表示 | EMDBヘッダ |
| FSC (解像度算出) | emd_25481_fsc.xml | 11.1 KB | 表示 | FSCデータファイル |
| 画像 |  emd_25481.png emd_25481.png | 105 KB | ||
| マスクデータ | emd_25481_msk_1.mapemd_25481_msk_2.map | 125 MB 125 MB | マスクマップ | |
| Filedesc metadata | emd-25481.cif.gz | 5.2 KB | ||
| その他 | emd_25481_additional_1.map.gzemd_25481_additional_2.map.gzemd_25481_additional_3.map.gzemd_25481_additional_4.map.gzemd_25481_half_map_1.map.gzemd_25481_half_map_2.map.gz | 33.6 MB 19.7 MB 64.4 MB 561.9 KB 115.9 MB 115.9 MB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-25481ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25481 http://ftp.pdbj.org/pub/emdb/structures/EMD-25481ftp://ftp.pdbj.org/pub/emdb/structures/EMD-25481 | HTTPS FTP |
-検証レポート
| 文書・要旨 | emd_25481_validation.pdf.gz | 770.1 KB | 表示 | EMDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | emd_25481_full_validation.pdf.gz | 769.6 KB | 表示 | |
| XML形式データ | emd_25481_validation.xml.gz | 19.1 KB | 表示 | |
| CIF形式データ | emd_25481_validation.cif.gz | 24.7 KB | 表示 | |
| アーカイブディレクトリ | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-25481ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-25481 | HTTPS FTP |
-関連構造データ
| 関連構造データ |  7swyFC  7tn2C C: 同じ文献を引用 ( F: あてはまる*YM |
|---|---|
| 類似構造データ | |
| 電子顕微鏡画像生データ | EMPIAR-10876 (タイトル: 2.3 A structure of the ATP-dependent chromatin remodeler Chd1 bound to the nucleosome in a nucleotide-free state Data size: 4.7 TB Data #1: Unaligned frames of Chd1-bound nucleosomes collected on Gatan K3 in Super Resolution [micrographs - multiframe] Data #2: MotionCor2-aligned frames of Chd1-bound nucleosomes collected on Gatan K3 [micrographs - single frame] Data #3: Processed subsets [picked particles - single frame - processed]) |












-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| 「今月の分子」の関連する項目 |


-マップ
| ファイル | ダウンロード / ファイル: emd_25481.map.gz / 形式: CCP4 / 大きさ: 125 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Sharpened 2.6A map in an orientation aligned to a common reference. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
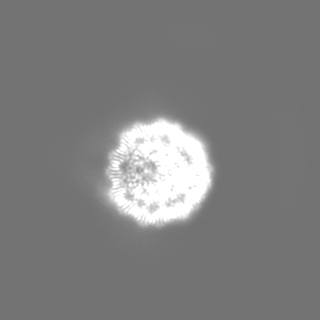
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 0.972 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-添付データ
-マスク #1
| ファイル | emd_25481_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
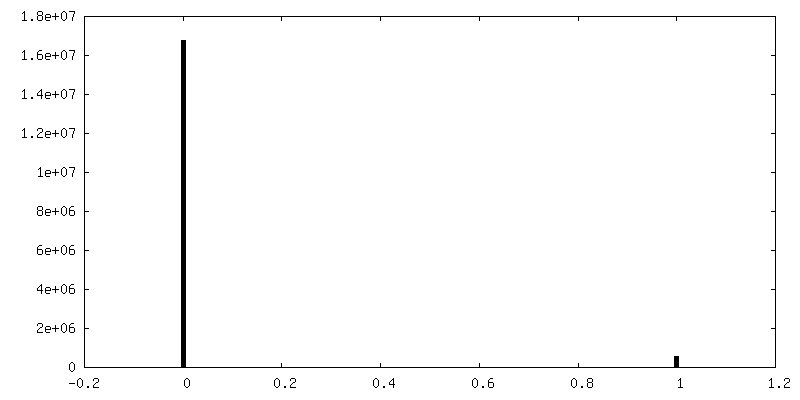
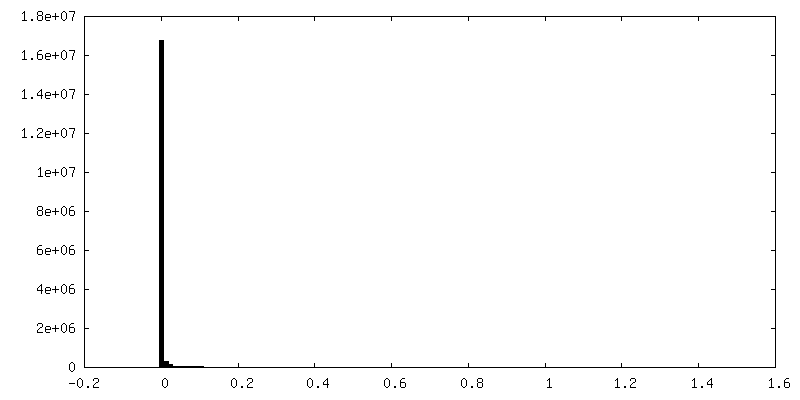
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |







-マスク #2
| ファイル | emd_25481_msk_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
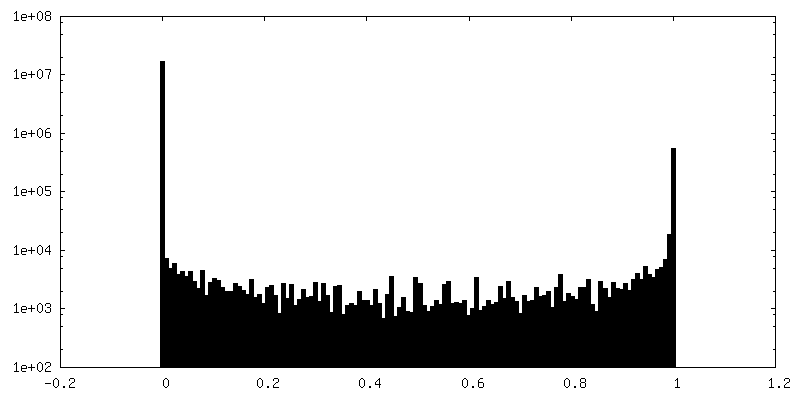
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |






-追加マップ: Unsharpened 2.6A map in an orientation aligned to a common reference.
| ファイル | emd_25481_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened 2.6A map in an orientation aligned to a common reference. | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |





-追加マップ: Unsharpened 2.6A map
| ファイル | emd_25481_additional_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Unsharpened 2.6A map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |





-追加マップ: Sharpened 2.6A map
| ファイル | emd_25481_additional_3.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | Sharpened 2.6A map | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |



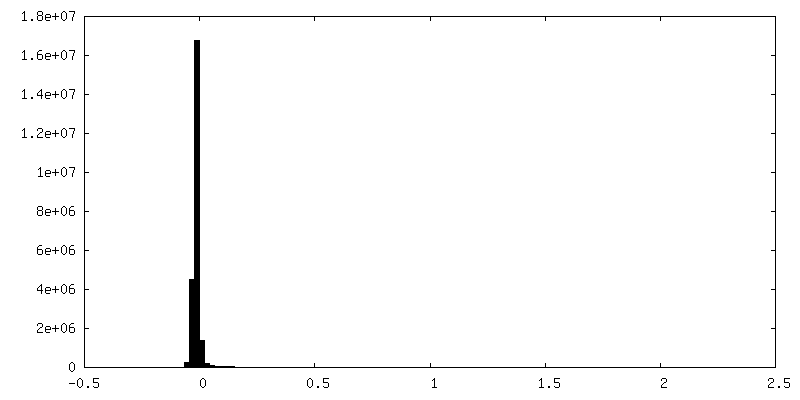
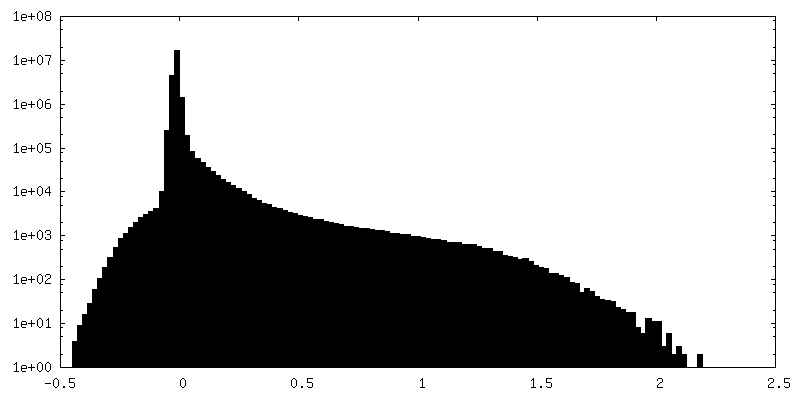
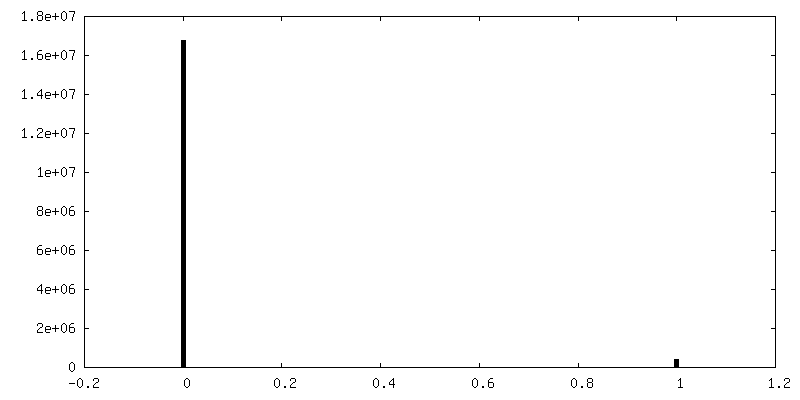
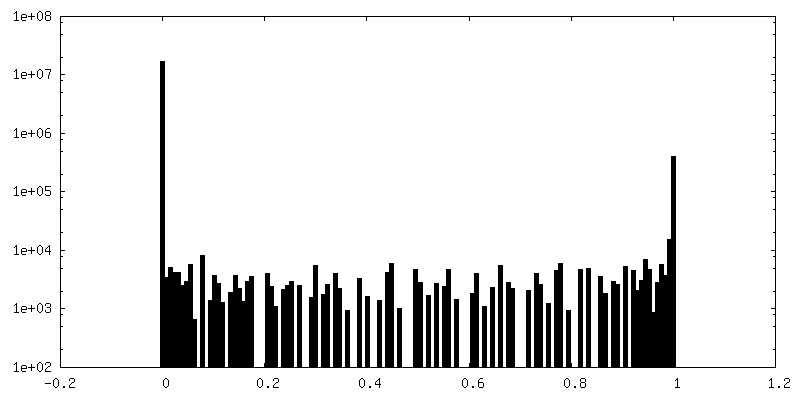
-追加マップ: FSC Mask from cryoSPARC (before auto-tightening)
| ファイル | emd_25481_additional_4.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | FSC Mask from cryoSPARC (before auto-tightening) | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |






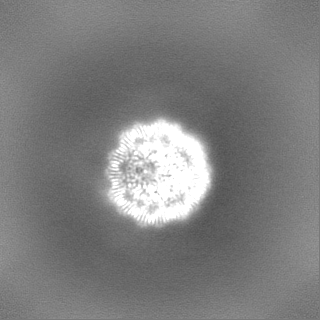
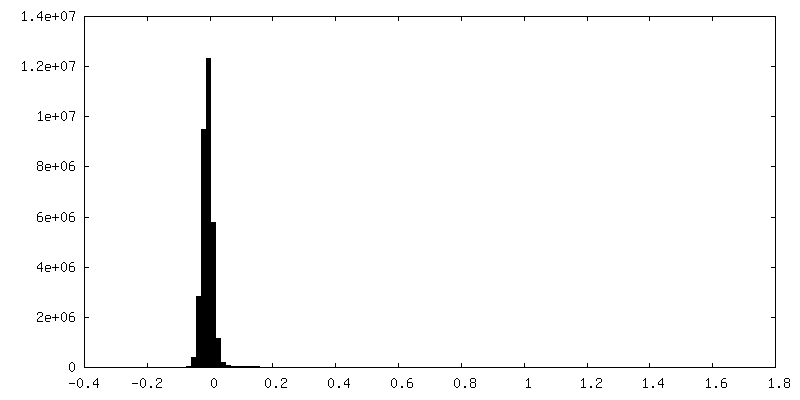
-ハーフマップ: half-map 1 (half A) from cryoSPARC
| ファイル | emd_25481_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half-map 1 (half_A) from cryoSPARC | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |



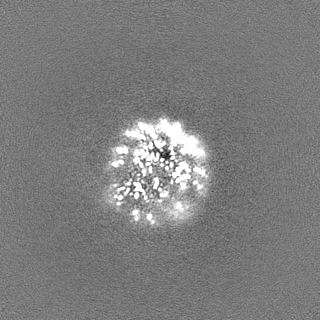
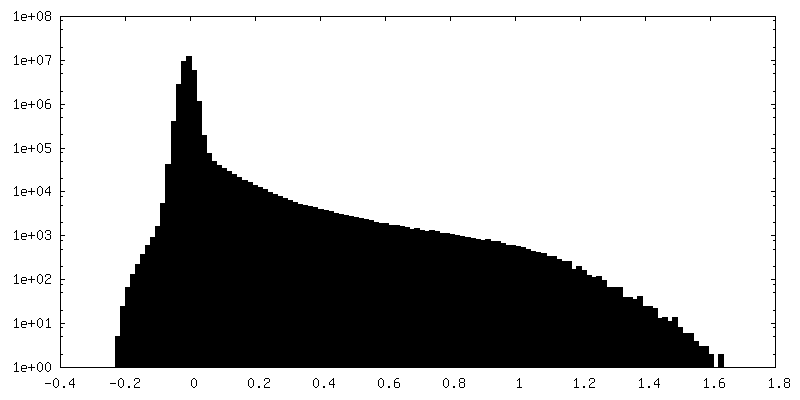
-ハーフマップ: half-map 2 (half B) from cryoSPARC
| ファイル | emd_25481_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 注釈 | half-map 2 (half_B) from cryoSPARC | ||||||||||||
| 投影像・断面図 |
| ||||||||||||
| 密度ヒストグラム |





- 試料の構成要素
試料の構成要素
-全体 : Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
| 全体 | 名称: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state |
|---|---|
| 要素 |
|
-超分子 #1: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
| 超分子 | 名称: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state タイプ: complex / ID: 1 / 親要素: 0 / 詳細: This entry has a weak DNA-binding domain |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 400 KDa |
-実験情報
-構造解析
| 手法 | クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 濃度 | 1.7 mg/mL |
|---|---|
| 緩衝液 | pH: 7 詳細: 20 mM HEPES, pH 7.0, 60 mM KCl, 1.5 mM DTT, 1 mM MgCl2 |
| グリッド | モデル: Quantifoil R2/2 / 材質: COPPER / メッシュ: 200 / 支持フィルム - 材質: CARBON / 支持フィルム - トポロジー: HOLEY |
| 凍結 | 凍結剤: ETHANE / チャンバー内湿度: 100 % / チャンバー内温度: 277 K / 装置: FEI VITROBOT MARK IV |
| 詳細 | Nucleosome-bound CHD1, prepared in the presence of ATPgammaS, and then crosslinked using GraFix. |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TITAN KRIOS |
|---|---|
| 特殊光学系 | エネルギーフィルター - スリット幅: 20 eV |
| 撮影 | フィルム・検出器のモデル: GATAN K3 (6k x 4k) / デジタル化 - サイズ - 横: 11520 pixel / デジタル化 - サイズ - 縦: 8184 pixel / 撮影したグリッド数: 1 / 実像数: 6906 / 平均露光時間: 3.3 sec. / 平均電子線量: 49.9 e/Å2 |
| 電子線 | 加速電圧: 300 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | C2レンズ絞り径: 100.0 µm / 最大 デフォーカス(補正後): 28.0 µm / 最小 デフォーカス(補正後): 4.0 µm / 倍率(補正後): 46296 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.7 mm / 最大 デフォーカス(公称値): -2.2 µm / 最小 デフォーカス(公称値): -1.0 µm / 倍率(公称値): 81000 |
| 試料ステージ | 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER ホルダー冷却材: NITROGEN |
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |