- EMDB-25480: 2.7 A structure of the ATP-dependent chromatin remodeler Chd1 bou... -
+
データを開く
IDまたはキーワード:
読み込み中...
-
基本情報
登録情報
データベース: EMDB / ID: EMD-25480
タイトル
2.7 A structure of the ATP-dependent chromatin remodeler Chd1 bound to the nucleosome in a nucleotide-free state. This entry contains a better resolved DNA-binding domain
マップデータ
Sharpened 2.7A map with a negative bfactor (-17) in an orientation aligned to a common reference
試料
複合体: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state
キーワード
CHD1 / chromatin remodeling / ATPase / DBD / nucleosome / remodeling / transcription / DNA BINDING PROTEIN
機能・相同性
機能・相同性情報
nucleolar chromatin / regulation of transcriptional start site selection at RNA polymerase II promoter / negative regulation of DNA-templated DNA replication / regulation of chromatin organization / SLIK (SAGA-like) complex / rDNA binding / DNA double-strand break processing / nucleosome organization / ATP-dependent chromatin remodeler activity / SAGA complex ...nucleolar chromatin / regulation of transcriptional start site selection at RNA polymerase II promoter / negative regulation of DNA-templated DNA replication / regulation of chromatin organization / SLIK (SAGA-like) complex / rDNA binding / DNA double-strand break processing / nucleosome organization / ATP-dependent chromatin remodeler activity / SAGA complex / sister chromatid cohesion / termination of RNA polymerase II transcription / termination of RNA polymerase I transcription / ATP-dependent activity, acting on DNA / : / transcription elongation by RNA polymerase II / helicase activity / double-strand break repair via homologous recombination / chromatin DNA binding / 加水分解酵素; 酸無水物に作用; 酸無水物に作用・細胞または細胞小器官の運動に関与 / structural constituent of chromatin / nucleosome / heterochromatin formation / nucleosome assembly / site of double-strand break / histone binding / transcription cis-regulatory region binding / chromatin remodeling / protein heterodimerization activity / chromatin binding / regulation of transcription by RNA polymerase II / chromatin / ATP hydrolysis activity / mitochondrion / DNA binding / ATP binding / nucleus 類似検索 - 分子機能
National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01-GM084192
米国
引用
ジャーナル: Nat Struct Mol Biol / 年: 2022 タイトル: Nucleosome recognition and DNA distortion by the Chd1 remodeler in a nucleotide-free state. 著者: Ilana M Nodelman / Sayan Das / Anneliese M Faustino / Stephen D Fried / Gregory D Bowman / Jean-Paul Armache / 要旨: Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free ...Chromatin remodelers are ATP-dependent enzymes that reorganize nucleosomes within all eukaryotic genomes. Here we report a complex of the Chd1 remodeler bound to a nucleosome in a nucleotide-free state, determined by cryo-EM to 2.3 Å resolution. The remodeler stimulates the nucleosome to absorb an additional nucleotide on each strand at two different locations: on the tracking strand within the ATPase binding site and on the guide strand one helical turn from the ATPase motor. Remarkably, the additional nucleotide on the tracking strand is associated with a local transformation toward an A-form geometry, explaining how sequential ratcheting of each DNA strand occurs. The structure also reveals a histone-binding motif, ChEx, which can block opposing remodelers on the nucleosome and may allow Chd1 to participate in histone reorganization during transcription.
EMPIAR-10876 (タイトル: 2.3 A structure of the ATP-dependent chromatin remodeler Chd1 bound to the nucleosome in a nucleotide-free state Data size: 4.7 TB Data #1: Unaligned frames of Chd1-bound nucleosomes collected on Gatan K3 in Super Resolution [micrographs - multiframe] Data #2: MotionCor2-aligned frames of Chd1-bound nucleosomes collected on Gatan K3 [micrographs - single frame] Data #3: Processed subsets [picked particles - single frame - processed])
全体 : Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
全体
名称: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state
要素
複合体: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state
-
超分子 #1: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a...
超分子
名称: Chd1 ATP-dependent chromatin remodeler bound to a nucleosome in a nucleotide-free state タイプ: complex / ID: 1 / 親要素: 0 / 詳細: This entry has a weak DNA-binding domain
由来(天然)
生物種: Saccharomyces cerevisiae (パン酵母)
分子量
理論値: 400 KDa
-
実験情報
-
構造解析
手法
クライオ電子顕微鏡法
解析
単粒子再構成法
試料の集合状態
particle
-
試料調製
濃度
1.7 mg/mL
緩衝液
pH: 7 詳細: 20 mM HEPES, pH 7.0, 60 mM KCl, 1.5 mM DTT, 1 mM MgCl2
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 マップデータ
マップデータ 試料
試料 キーワード
キーワード 機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 米国, 1件
米国, 1件  引用
引用 構造の表示
構造の表示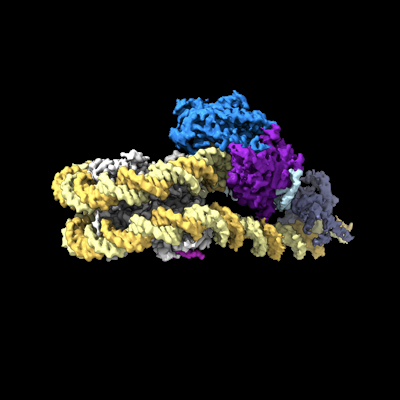
 ダウンロードとリンク
ダウンロードとリンク emd_25480.png
emd_25480.png http://ftp.pdbj.org/pub/emdb/structures/EMD-25480
http://ftp.pdbj.org/pub/emdb/structures/EMD-25480















 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)













































































 試料の構成要素
試料の構成要素 解析
解析 電子顕微鏡法
電子顕微鏡法 FIELD EMISSION GUN
FIELD EMISSION GUN



