Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-23356 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
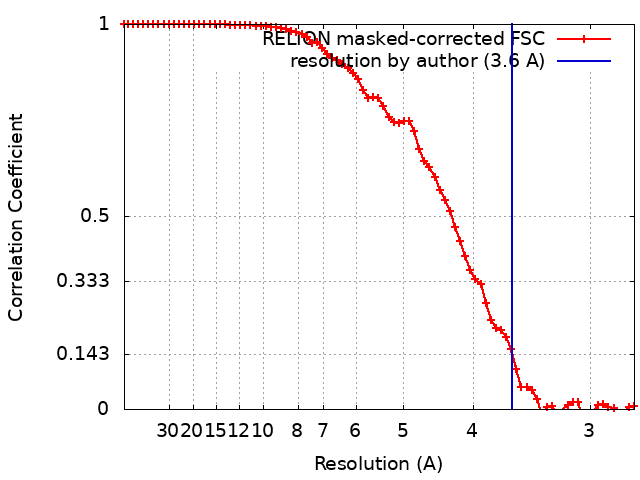
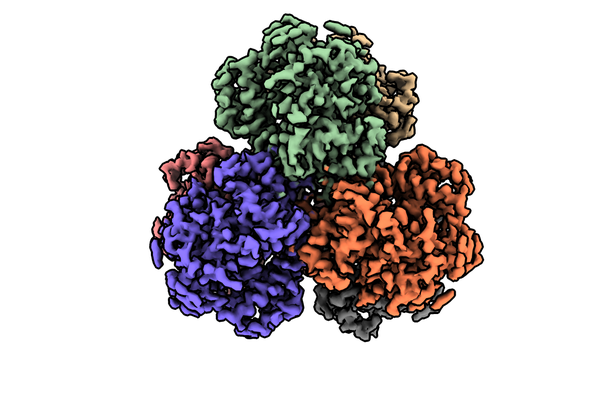
| Title | Structure of dNTPase at 3.6 Angstrom Resolution | |||||||||
 Map data Map data | Structure of dNTPase at 3.6 Angstrom Resolution | |||||||||
 Sample Sample |
| |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |   Vibrio cholerae (bacteria) Vibrio cholerae (bacteria) | |||||||||
| Method | subtomogram averaging / cryo EM / Resolution: 3.6 Å | |||||||||
 Authors Authors | Bouvette J / Liu HF / Du X / Zhou Y / Sikkema AP / Mello JFR / Klemm B / Huang R / Schaaper RM / Borgnia MJ / Bartesaghi A | |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2021 Title: Beam image-shift accelerated data acquisition for near-atomic resolution single-particle cryo-electron tomography. Authors: Jonathan Bouvette / Hsuan-Fu Liu / Xiaochen Du / Ye Zhou / Andrew P Sikkema / Juliana da Fonseca Rezende E Mello / Bradley P Klemm / Rick Huang / Roel M Schaaper / Mario J Borgnia / Alberto Bartesaghi /  Abstract: Tomographic reconstruction of cryopreserved specimens imaged in an electron microscope followed by extraction and averaging of sub-volumes has been successfully used to derive atomic models of ...Tomographic reconstruction of cryopreserved specimens imaged in an electron microscope followed by extraction and averaging of sub-volumes has been successfully used to derive atomic models of macromolecules in their biological environment. Eliminating biochemical isolation steps required by other techniques, this method opens up the cell to in-situ structural studies. However, the need to compensate for errors in targeting introduced during mechanical navigation of the specimen significantly slows down tomographic data collection thus limiting its practical value. Here, we introduce protocols for tilt-series acquisition and processing that accelerate data collection speed by up to an order of magnitude and improve map resolution compared to existing approaches. We achieve this by using beam-image shift to multiply the number of areas imaged at each stage position, by integrating geometrical constraints during imaging to achieve high precision targeting, and by performing per-tilt astigmatic CTF estimation and data-driven exposure weighting to improve final map resolution. We validated our beam image-shift electron cryo-tomography (BISECT) approach by determining the structure of a low molecular weight target (~300 kDa) at 3.6 Å resolution where density for individual side chains is clearly resolved. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_23356.map.gz | 2.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-23356-v30.xmlemd-23356.xml | 16.3 KB 16.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_23356_fsc.xml | 7.2 KB | Display | FSC data file |
| Images | 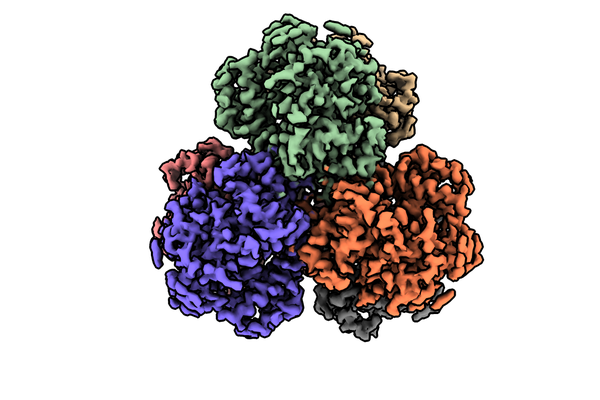 emd_23356.png emd_23356.png | 175.5 KB | ||
| Masks | emd_23356_msk_1.map | 30.5 MB | Mask map | |
| Others | emd_23356_additional_1.map.gzemd_23356_half_map_1.map.gzemd_23356_half_map_2.map.gz | 28 MB 16.5 MB 16.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-23356ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23356 http://ftp.pdbj.org/pub/emdb/structures/EMD-23356ftp://ftp.pdbj.org/pub/emdb/structures/EMD-23356 | HTTPS FTP |
-Related structure data
| Related structure data |  7lw5 M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_23356.map.gz / Format: CCP4 / Size: 2.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of dNTPase at 3.6 Angstrom Resolution | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.37 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_23356_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
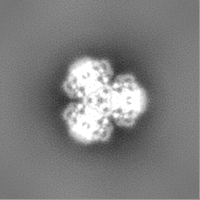
| Projections & Slices |
| ||||||||||||
| Density Histograms |






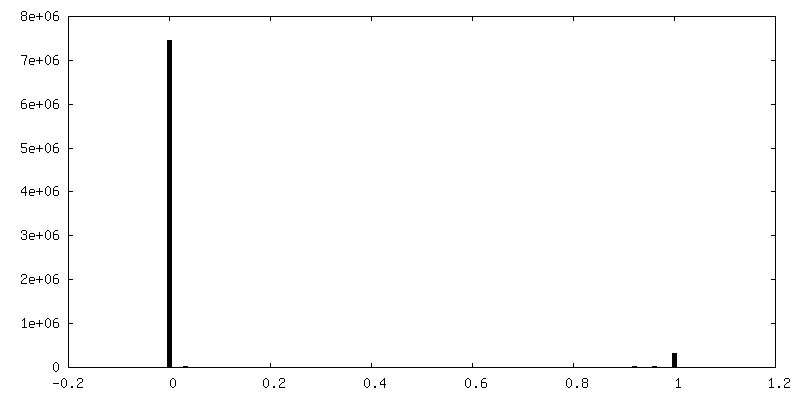
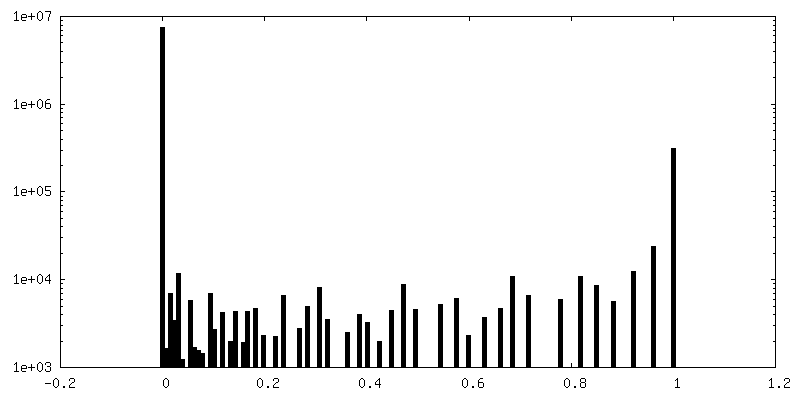
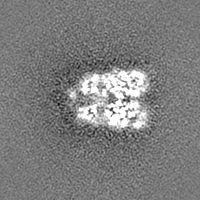
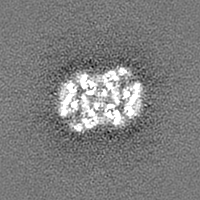
-Additional map: Structure of dNTPase at 3.6 Angstrom Resolution
| File | emd_23356_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of dNTPase at 3.6 Angstrom Resolution | ||||||||||||
| Projections & Slices |
| ||||||||||||
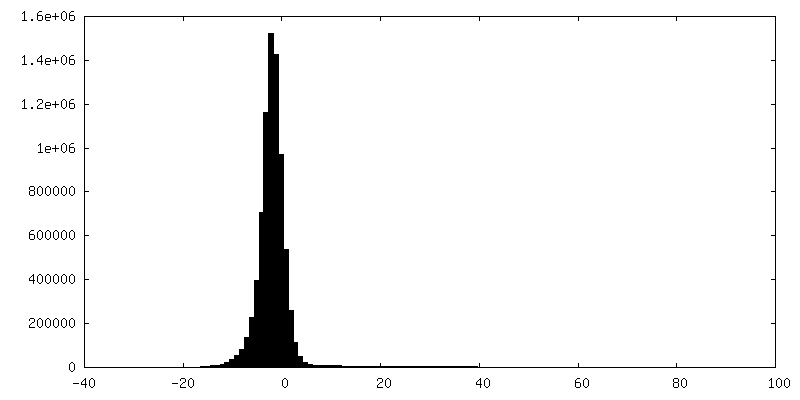
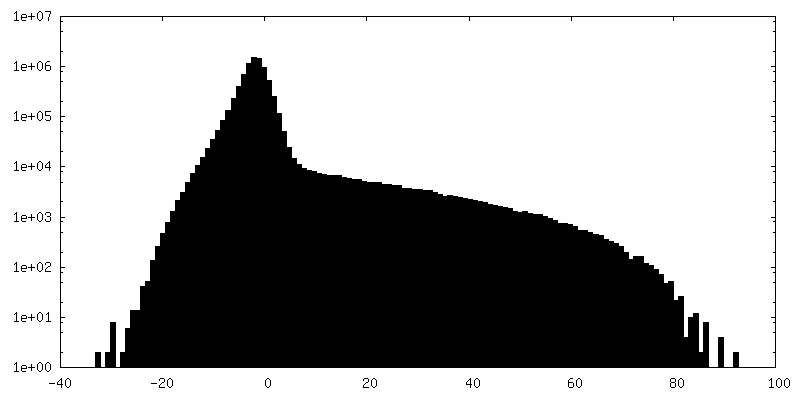
| Density Histograms |

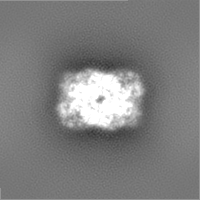

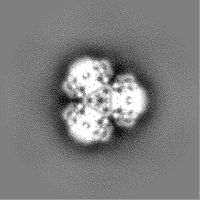
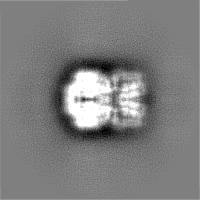
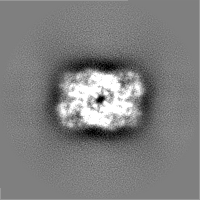
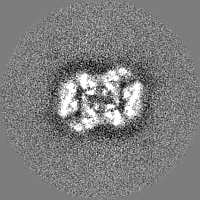
-Half map: Structure of dNTPase at 3.6 Angstrom Resolution
| File | emd_23356_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of dNTPase at 3.6 Angstrom Resolution | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


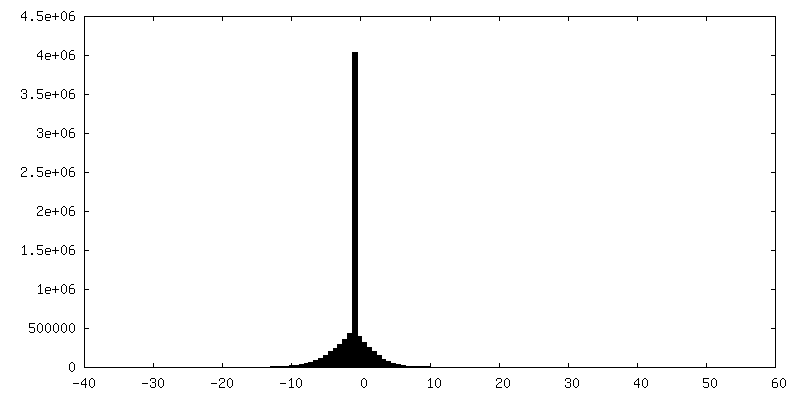
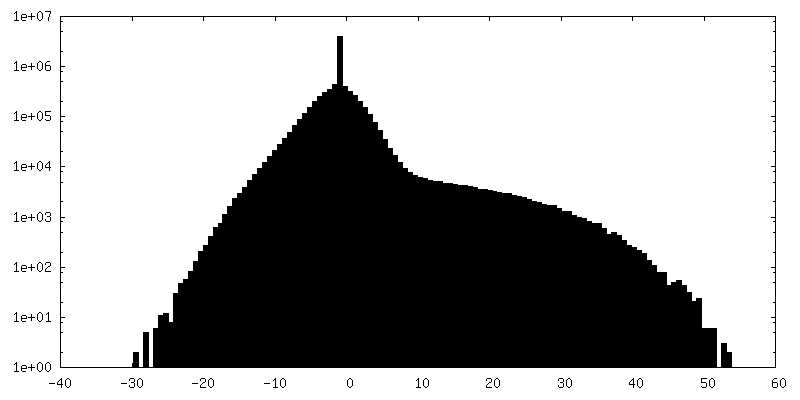
-Half map: Structure of dNTPase at 3.6 Angstrom Resolution
| File | emd_23356_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Structure of dNTPase at 3.6 Angstrom Resolution | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |
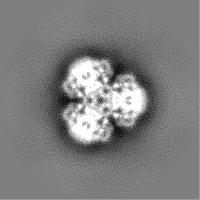
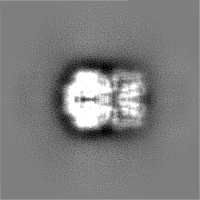




- Sample components
Sample components
-Entire : dNTPase
| Entire | Name: dNTPase |
|---|---|
| Components |
|
-Supramolecule #1: dNTPase
| Supramolecule | Name: dNTPase / type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Vibrio cholerae (bacteria) |
| Recombinant expression | Organism: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | subtomogram averaging |
| Aggregation state | particle |
-Sample preparation
| Concentration | 1.3 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Pretreatment - Type: GLOW DISCHARGE |
| Vitrification | Cryogen name: ETHANE / Instrument: LEICA EM GP |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Average electron dose: 5.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |