oxidation-dependent protein catabolic process / response to aluminum ion / PH domain binding / mitochondrial protein catabolic process / endopeptidase La / G-quadruplex DNA binding / mitochondrial DNA metabolic process / : / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins ...oxidation-dependent protein catabolic process / response to aluminum ion / PH domain binding / mitochondrial protein catabolic process / endopeptidase La / G-quadruplex DNA binding / mitochondrial DNA metabolic process / : / ATP-dependent peptidase activity / protein quality control for misfolded or incompletely synthesized proteins / mitochondrial nucleoid / insulin receptor substrate binding / Mitochondrial unfolded protein response (UPRmt) / chaperone-mediated protein complex assembly / response to hormone / DNA polymerase binding / Mitochondrial protein degradation / negative regulation of insulin receptor signaling pathway / proteolysis involved in protein catabolic process / mitochondrion organization / protein catabolic process / ADP binding / single-stranded DNA binding / cellular response to oxidative stress / sequence-specific DNA binding / response to hypoxia / single-stranded RNA binding / mitochondrial matrix / serine-type endopeptidase activity / ATP hydrolysis activity / mitochondrion / nucleoplasm / ATP binding / identical protein binding / membrane / cytosol Similarity search - Function
Lon protease homologue, chloroplastic/mitochondrial / : / Lon protease, bacterial/eukaryotic-type / Lon protease AAA+ ATPase lid domain / Peptidase S16, active site / ATP-dependent serine proteases, lon family, serine active site. / Lon proteolytic domain profile. / Peptidase S16, Lon proteolytic domain / Lon protease / Lon protease (S16) C-terminal proteolytic domain ...Lon protease homologue, chloroplastic/mitochondrial / : / Lon protease, bacterial/eukaryotic-type / Lon protease AAA+ ATPase lid domain / Peptidase S16, active site / ATP-dependent serine proteases, lon family, serine active site. / Lon proteolytic domain profile. / Peptidase S16, Lon proteolytic domain / Lon protease / Lon protease (S16) C-terminal proteolytic domain / Lon N-terminal domain profile. / Lon protease, N-terminal domain / Lon protease, N-terminal domain superfamily / ATP-dependent protease La (LON) substrate-binding domain / Found in ATP-dependent protease La (LON) / PUA-like superfamily / ATPase family associated with various cellular activities (AAA) / ATPase, AAA-type, core / Ribosomal protein S5 domain 2-type fold, subgroup / Ribosomal protein S5 domain 2-type fold / ATPases associated with a variety of cellular activities / AAA+ ATPase domain / P-loop containing nucleoside triphosphate hydrolase Similarity search - Domain/homology
National Institutes of Health/National Institute on Aging (NIH/NIA)
AG067594
United States
National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS)
NS095892
United States
National Institutes of Health/National Institute on Aging (NIH/NIA)
AG061697
United States
National Institutes of Health/Office of the Director
S10OD021634
United States
Citation
Journal: Nat Commun / Year: 2021 Title: Structures of the human LONP1 protease reveal regulatory steps involved in protease activation. Authors: Mia Shin / Edmond R Watson / Albert S Song / Jeffrey T Mindrebo / Scott J Novick / Patrick R Griffin / R Luke Wiseman / Gabriel C Lander / Abstract: The human mitochondrial AAA+ protein LONP1 is a critical quality control protease involved in regulating diverse aspects of mitochondrial biology including proteostasis, electron transport chain ...The human mitochondrial AAA+ protein LONP1 is a critical quality control protease involved in regulating diverse aspects of mitochondrial biology including proteostasis, electron transport chain activity, and mitochondrial transcription. As such, genetic or aging-associated imbalances in LONP1 activity are implicated in pathologic mitochondrial dysfunction associated with numerous human diseases. Despite this importance, the molecular basis for LONP1-dependent proteolytic activity remains poorly defined. Here, we solved cryo-electron microscopy structures of human LONP1 to reveal the underlying molecular mechanisms governing substrate proteolysis. We show that, like bacterial Lon, human LONP1 adopts both an open and closed spiral staircase orientation dictated by the presence of substrate and nucleotide. Unlike bacterial Lon, human LONP1 contains a second spiral staircase within its ATPase domain that engages substrate as it is translocated toward the proteolytic chamber. Intriguingly, and in contrast to its bacterial ortholog, substrate binding within the central ATPase channel of LONP1 alone is insufficient to induce the activated conformation of the protease domains. To successfully induce the active protease conformation in substrate-bound LONP1, substrate binding within the protease active site is necessary, which we demonstrate by adding bortezomib, a peptidomimetic active site inhibitor of LONP1. These results suggest LONP1 can decouple ATPase and protease activities depending on whether AAA+ or both AAA+ and protease domains bind substrate. Importantly, our structures provide a molecular framework to define the critical importance of LONP1 in regulating mitochondrial proteostasis in health and disease.
History
Deposition
Nov 23, 2020
-
Header (metadata) release
Dec 2, 2020
-
Map release
Dec 2, 2020
-
Update
May 29, 2024
-
Current status
May 29, 2024
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Details: Solutions were made fresh from concentrated and filtered using a 0.1 um syringe filter to avoid microbial contamination. Samples were mixed on ice and incubated for 5 minutes before vitrification.
Grid
Model: UltrAuFoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Time: 7 sec. / Pretreatment - Atmosphere: OTHER Details: EM grids were plasma cleaned prior to sample application for 7 seconds using a Solarus plasma cleaner (Gatan, Inc.) with a 75% nitrogen, 25% oxygen atmosphere at 15W.
Vitrification
Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: HOMEMADE PLUNGER Details: 4 uL of sample was applied per grid and manually blotted for 4 seconds followed by immediately plunge-freezing in liquid ethane cooled by liquid nitrogen..
Details
This sample was monodisperse
-
Electron microscopy
Microscope
FEI TALOS ARCTICA
Temperature
Min: 80.0 K / Max: 90.0 K
Alignment procedure
Coma free - Residual tilt: 0.14 mrad
Details
Coma-free alignment procedure from Herzik & Wu, Nature Methods (2017). Preliminary grid screening was performed manually prior to data collection.
Image recording
Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3710 pixel / Digitization - Dimensions - Height: 3838 pixel / Digitization - Frames/image: 0-113 / Number grids imaged: 1 / Number real images: 2912 / Average exposure time: 11.4 sec. / Average electron dose: 50.0 e/Å2 Details: Images were collected in counting mode at 10 frames per second.
Electron beam
Acceleration voltage: 200 kV / Electron source: FIELD EMISSION GUN
Model: Talos Arctica / Image courtesy: FEI Company
+
Image processing
Particle selection
Number selected: 940396
Startup model
Type of model: OTHER Details: A low-resolution negative stain reconstruction of Human mitochondrial LONP1 was used as an initial model.
Final reconstruction
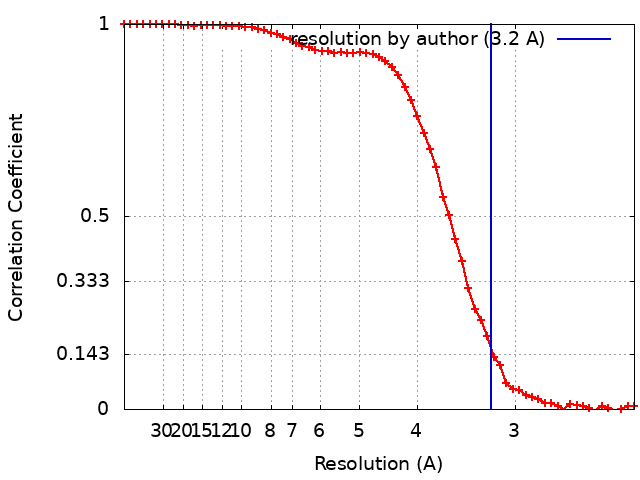
Number classes used: 1 / Resolution.type: BY AUTHOR / Resolution: 3.2 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 3.1) Software - details: RELION 3.1 was used to perform final reconstruction Number images used: 38130
Initial angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) Software - details: RELION 3.1 was used to assign initial euler angles
Final angle assignment
Type: MAXIMUM LIKELIHOOD / Software - Name: RELION (ver. 3.1) Software - details: RELION 3.1 was used to assign final euler angles Details: RELION 3.1 was used to assign initial angles
Final 3D classification
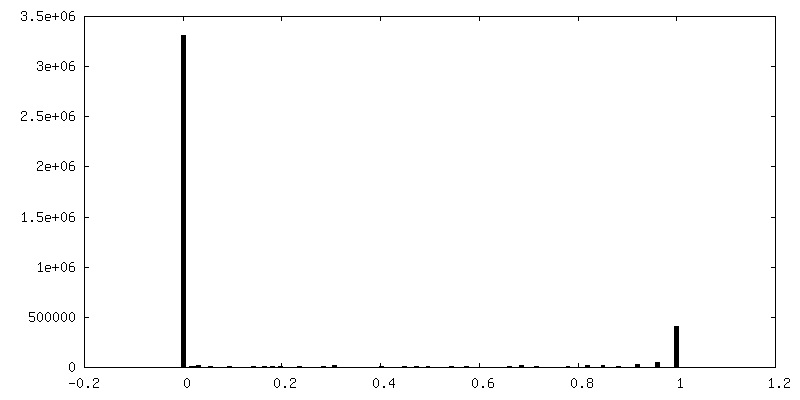
Number classes: 3 / Avg.num./class: 90000 / Software - Name: RELION (ver. 3.1) Software - details: RELION 3.1 was used to perform final classification Details: The final 3D classification had an somewhat asymmetric distribution owing to preferred specimen orientation due to differential interactions with the air-water interface
FSC plot (resolution estimation)
-
Atomic model buiding 1
Details
Initial homology model was built using SWISS-MODEL and initial rigid body docking was done using UCSF Chimera.
Refinement
Space: REAL / Protocol: AB INITIO MODEL / Overall B value: 52 / Target criteria: Correlation coefficient
Output model
PDB-7ksm: Human mitochondrial LONP1 with endogenous substrate
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human) /
Homo sapiens (human) / 
 Authors
Authors United States, 4 items
United States, 4 items  Citation
Citation Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_23020.png
emd_23020.png http://ftp.pdbj.org/pub/emdb/structures/EMD-23020
http://ftp.pdbj.org/pub/emdb/structures/EMD-23020












 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)







































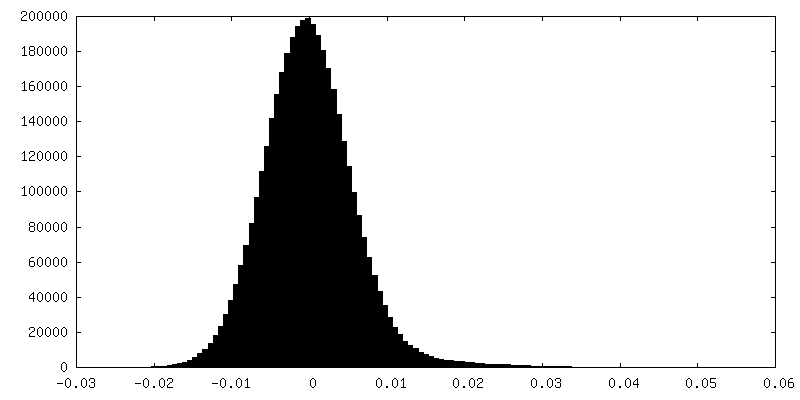

 Sample components
Sample components

 Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN