 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-2277: Ribosome structures to near-atomic resolution from thirty thousan... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-2277 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles | |||||||||
 Map data Map data | Reconstruction of 70S ribosome | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | cryo-EM / single-particle analysis / direct electron detectors / ribosome / RELION / statistical movie processing | |||||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 5.1 Å | |||||||||
 Authors Authors | Bai XC / Fernandez IS / McMullan G / Scheres SHW | |||||||||
 Citation Citation | Journal: Elife / Year: 2013 Title: Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. Authors: Xiao-Chen Bai / Israel S Fernandez / Greg McMullan / Sjors H W Scheres /  Abstract: Although electron cryo-microscopy (cryo-EM) single-particle analysis has become an important tool for structural biology of large and flexible macro-molecular assemblies, the technique has not yet ...Although electron cryo-microscopy (cryo-EM) single-particle analysis has become an important tool for structural biology of large and flexible macro-molecular assemblies, the technique has not yet reached its full potential. Besides fundamental limits imposed by radiation damage, poor detectors and beam-induced sample movement have been shown to degrade attainable resolutions. A new generation of direct electron detectors may ameliorate both effects. Apart from exhibiting improved signal-to-noise performance, these cameras are also fast enough to follow particle movements during electron irradiation. Here, we assess the potentials of this technology for cryo-EM structure determination. Using a newly developed statistical movie processing approach to compensate for beam-induced movement, we show that ribosome reconstructions with unprecedented resolutions may be calculated from almost two orders of magnitude fewer particles than used previously. Therefore, this methodology may expand the scope of high-resolution cryo-EM to a broad range of biological specimens.DOI:http://dx.doi.org/10.7554/eLife.00461.001. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_2277.map.gz | 28.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-2277-v30.xmlemd-2277.xml | 10.3 KB 10.3 KB | Display Display | EMDB header |
| Images | 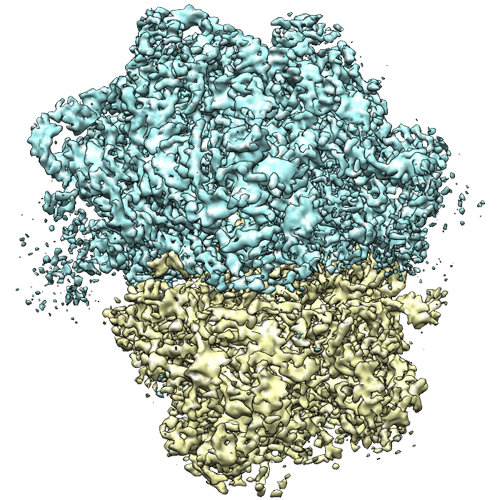 EMD-2277.jpg EMD-2277.jpg | 298.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-2277ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2277 http://ftp.pdbj.org/pub/emdb/structures/EMD-2277ftp://ftp.pdbj.org/pub/emdb/structures/EMD-2277 | HTTPS FTP |
-Related structure data












-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_2277.map.gz / Format: CCP4 / Size: 29.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Reconstruction of 70S ribosome | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.77 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : T. thermophilus 70S ribosome
| Entire | Name: T. thermophilus 70S ribosome |
|---|---|
| Components |
|
-Supramolecule #1000: T. thermophilus 70S ribosome
| Supramolecule | Name: T. thermophilus 70S ribosome / type: sample / ID: 1000 / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 3 MDa / Theoretical: 3 MDa |
-Supramolecule #1: 70S ribosome
| Supramolecule | Name: 70S ribosome / type: complex / ID: 1 / Recombinant expression: No / Ribosome-details: ribosome-prokaryote: ALL |
|---|---|
| Source (natural) | Organism: Thermus thermophilus (bacteria) |
| Molecular weight | Experimental: 3 MDa / Theoretical: 3 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL |
|---|---|
| Buffer | pH: 7.45 Details: 3mM Hepes-KOH, 6.6 mM Tris-acetate pH 7.2, 3 mM NH4Cl, 6.6 mM NH4-acetate, 48 mM K-acetate, 4 mM Mg-acetate, 2.4 mM DTT |
| Grid | Details: Quantifoil grids (2/2) with 3 nm thin carbon on top |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 90 K / Instrument: FEI VITROBOT MARK II / Method: Blot 2.5 seconds before plunging |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Temperature | Min: 80 K / Max: 90 K / Average: 85 K |
| Alignment procedure | Legacy - Astigmatism: Objective lens astigmatism was corrected at 59,000 times magnification |
| Date | Jul 1, 2012 |
| Image recording | Category: CCD / Film or detector model: FEI FALCON I (4k x 4k) / Digitization - Sampling interval: 14 µm / Number real images: 360 / Average electron dose: 16 e/Å2 Details: Every image is the average of sixteen frames recorded by the direct electron detector |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 79096 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 3.597 µm / Nominal defocus min: 1.178 µm / Nominal magnification: 59000 |
| Sample stage | Specimen holder model: GATAN LIQUID NITROGEN |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
-Image processing
| Details | Use a newly developed statistical movie processing to compensate for beam-induced movement |
|---|---|
| CTF correction | Details: each image |
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Resolution.type: BY AUTHOR / Resolution: 5.1 Å / Resolution method: OTHER / Software - Name: CTFFIND3, RELION Details: Use a newly developed statistical movie processing approach to compensate for beam-induced movement Number images used: 35404 |
-Atomic model buiding 1
| Initial model | PDB ID:  2wh1 |
|---|---|
| Software | Name: Chimera |
| Details | Protocol: Rigid body. The domains were separately fitted by manual docking using program Chimera |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
-Atomic model buiding 2
| Initial model | PDB ID: 2wh2 |
|---|---|
| Software | Name: Chimera |
| Details | Protocol: Rigid body. The domains were separately fitted by manual docking using program Chimera |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
