 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-22322: Cryo-EM structure of bedaquiline-washed Mycobacterium smegmatis A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22322 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Cryo-EM structure of bedaquiline-washed Mycobacterium smegmatis ATP synthase FO region | |||||||||
 Map data Map data | Locally sharpened map. | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  Mycolicibacterium smegmatis (bacteria) Mycolicibacterium smegmatis (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.7 Å | |||||||||
 Authors Authors | Guo H / Courbon GM / Rubinstein JL | |||||||||
| Funding support |  Canada, 2 items Canada, 2 items
| |||||||||
 Citation Citation | Journal: Nature / Year: 2021 Title: Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Authors: Hui Guo / Gautier M Courbon / Stephanie A Bueler / Juntao Mai / Jun Liu / John L Rubinstein / Abstract: Tuberculosis-the world's leading cause of death by infectious disease-is increasingly resistant to current first-line antibiotics. The bacterium Mycobacterium tuberculosis (which causes tuberculosis) ...Tuberculosis-the world's leading cause of death by infectious disease-is increasingly resistant to current first-line antibiotics. The bacterium Mycobacterium tuberculosis (which causes tuberculosis) can survive low-energy conditions, allowing infections to remain dormant and decreasing their susceptibility to many antibiotics. Bedaquiline was developed in 2005 from a lead compound identified in a phenotypic screen against Mycobacterium smegmatis. This drug can sterilize even latent M. tuberculosis infections and has become a cornerstone of treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Bedaquiline targets the mycobacterial ATP synthase, which is an essential enzyme in the obligate aerobic Mycobacterium genus, but how it binds the intact enzyme is unknown. Here we determined cryo-electron microscopy structures of M. smegmatis ATP synthase alone and in complex with bedaquiline. The drug-free structure suggests that hook-like extensions from the α-subunits prevent the enzyme from running in reverse, inhibiting ATP hydrolysis and preserving energy in hypoxic conditions. Bedaquiline binding induces large conformational changes in the ATP synthase, creating tight binding pockets at the interface of subunits a and c that explain the potency of this drug as an antibiotic for tuberculosis. #1: Journal: Biorxiv / Year: 2020Title: Structure of mycobacterial ATP synthase with the TB drug bedaquiline Authors: Guo H / Courbon GM / Bueler SA / Mai J / Liu J / Rubinstein JL | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22322.map.gz | 58.4 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22322-v30.xmlemd-22322.xml | 21.6 KB 21.6 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_22322_fsc.xml | 11.4 KB | Display | FSC data file |
| Images | 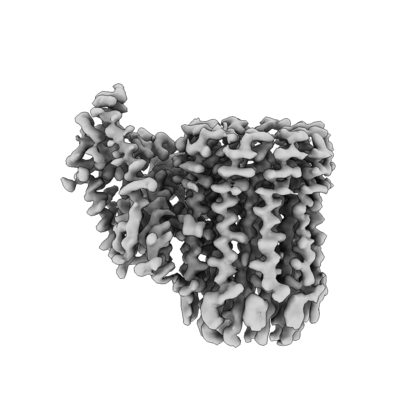 emd_22322.png emd_22322.png | 89.9 KB | ||
| Masks | emd_22322_msk_1.map | 125 MB | Mask map | |
| Others | emd_22322_additional.map.gzemd_22322_additional_1.map.gzemd_22322_half_map_1.map.gzemd_22322_half_map_2.map.gz | 61.5 MB 61.5 MB 115.9 MB 115.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22322ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22322 http://ftp.pdbj.org/pub/emdb/structures/EMD-22322ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22322 | HTTPS FTP |
-Related structure data
| Related structure data |  7jg5C  7jg6C  7jg7C  7jg8C  7jg9C  7jgaC  7jgbC  7jgcC C: citing same article ( |
|---|---|
| Similar structure data |

















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_22322.map.gz / Format: CCP4 / Size: 125 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Locally sharpened map. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.03 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Mask #1
| File | emd_22322_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
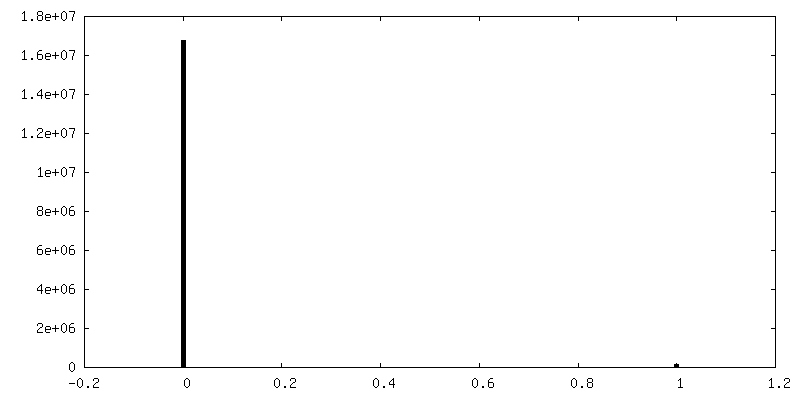
| Density Histograms |







-Additional map: Unsharpened map.
| File | emd_22322_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened map. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |






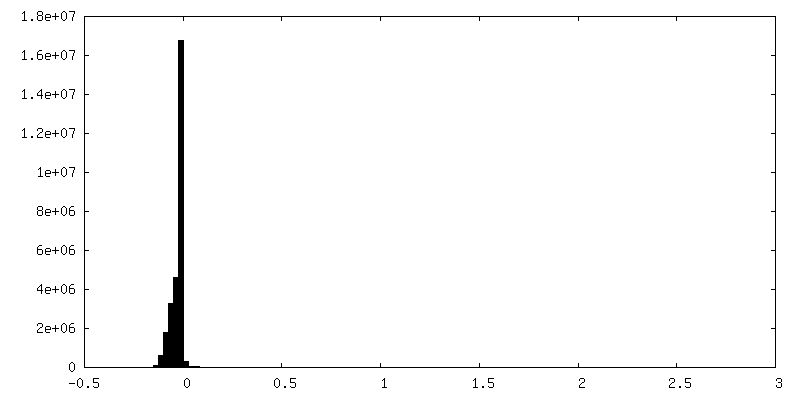

-Additional map: Unsharpened map.
| File | emd_22322_additional_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unsharpened map. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




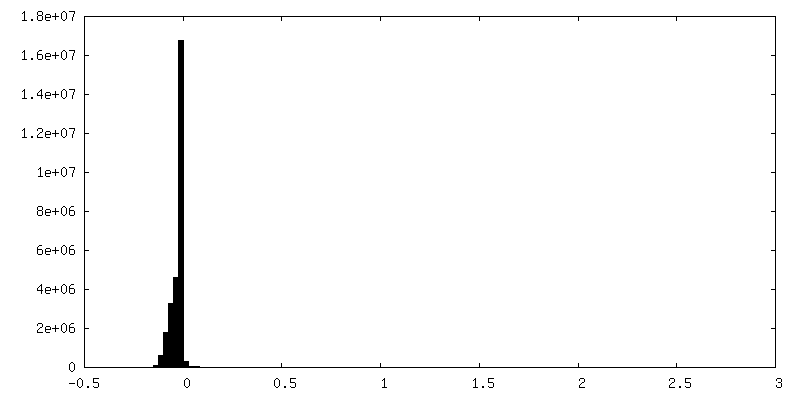
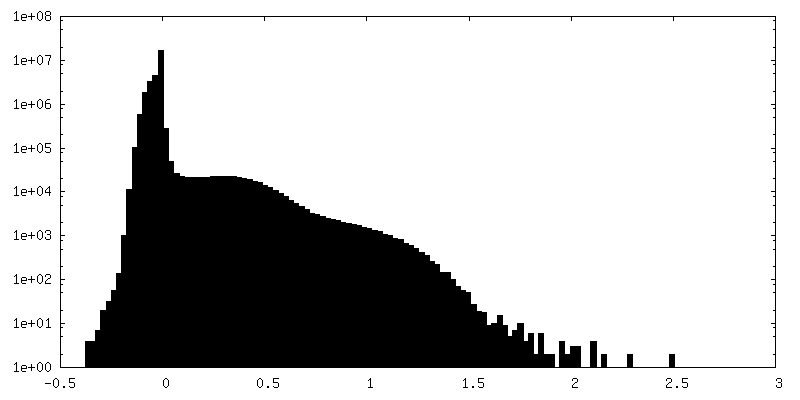
-Half map: Half map 1.
| File | emd_22322_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 1. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




-Half map: Half map 2.
| File | emd_22322_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half map 2. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




- Sample components
Sample components
-Entire : FO region of ATP synthase from Mycobacterium smegmatis
| Entire | Name: FO region of ATP synthase from Mycobacterium smegmatis |
|---|---|
| Components |
|
-Supramolecule #1: FO region of ATP synthase from Mycobacterium smegmatis
| Supramolecule | Name: FO region of ATP synthase from Mycobacterium smegmatis type: complex / ID: 1 / Parent: 0 |
|---|---|
| Source (natural) | Organism: Mycolicibacterium smegmatis (bacteria) |
| Molecular weight | Theoretical: 110 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 6 mg/mL |
|---|---|
| Buffer | pH: 7.4 |
| Grid | Model: Homemade / Material: COPPER/RHODIUM / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Atmosphere: OTHER |
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK III |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: FEI FALCON IV (4k x 4k) / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number real images: 4962 / Average electron dose: 41.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Calibrated defocus max: 4.0 µm / Calibrated defocus min: 0.5 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Refinement | Space: RECIPROCAL / Protocol: AB INITIO MODEL |
|---|
