 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-20629: Best fitting antiparallel model for Volume 2 of truncated dimeric... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-20629 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Best fitting antiparallel model for Volume 2 of truncated dimeric Cytohesin-3 (Grp1; amino acids 14-399) | |||||||||
 Map data Map data | Volume 2 for truncated dimeric Cytohesin-3 (Grp1; amino acids 14-399) | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Arf GEF / Phosphoinositide binding / Sec7 Domain / PH Domain / ENDOCYTOSIS | |||||||||
| Function / homology |  Function and homology information Function and homology informationestablishment of epithelial cell polarity / Golgi vesicle transport / Intra-Golgi traffic / regulation of ARF protein signal transduction / phosphatidylinositol-3,4,5-trisphosphate binding / bicellular tight junction / ruffle / positive regulation of cell adhesion / guanyl-nucleotide exchange factor activity / adherens junction ...establishment of epithelial cell polarity / Golgi vesicle transport / Intra-Golgi traffic / regulation of ARF protein signal transduction / phosphatidylinositol-3,4,5-trisphosphate binding / bicellular tight junction / ruffle / positive regulation of cell adhesion / guanyl-nucleotide exchange factor activity / adherens junction / Golgi membrane / nucleoplasm / plasma membrane / cytosol Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 53.0 Å | |||||||||
 Authors Authors | Das S / Lambright DG | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2019 Title: Structural Organization and Dynamics of Homodimeric Cytohesin Family Arf GTPase Exchange Factors in Solution and on Membranes. Authors: Sanchaita Das / Andrew W Malaby / Agata Nawrotek / Wenhua Zhang / Mahel Zeghouf / Sarah Maslen / Mark Skehel / Srinivas Chakravarthy / Thomas C Irving / Osman Bilsel / Jacqueline Cherfils / ...Authors: Sanchaita Das / Andrew W Malaby / Agata Nawrotek / Wenhua Zhang / Mahel Zeghouf / Sarah Maslen / Mark Skehel / Srinivas Chakravarthy / Thomas C Irving / Osman Bilsel / Jacqueline Cherfils / David G Lambright /   Abstract: Membrane dynamic processes require Arf GTPase activation by guanine nucleotide exchange factors (GEFs) with a Sec7 domain. Cytohesin family Arf GEFs function in signaling and cell migration through ...Membrane dynamic processes require Arf GTPase activation by guanine nucleotide exchange factors (GEFs) with a Sec7 domain. Cytohesin family Arf GEFs function in signaling and cell migration through Arf GTPase activation on the plasma membrane and endosomes. In this study, the structural organization of two cytohesins (Grp1 and ARNO) was investigated in solution by size exclusion-small angle X-ray scattering and negative stain-electron microscopy and on membranes by dynamic light scattering, hydrogen-deuterium exchange-mass spectrometry and guanosine diphosphate (GDP)/guanosine triphosphate (GTP) exchange assays. The results suggest that cytohesins form elongated dimers with a central coiled coil and membrane-binding pleckstrin-homology (PH) domains at opposite ends. The dimers display significant conformational heterogeneity, with a preference for compact to intermediate conformations. Phosphoinositide-dependent membrane recruitment is mediated by one PH domain at a time and alters the conformational dynamics to prime allosteric activation by Arf-GTP. A structural model for membrane targeting and allosteric activation of full-length cytohesin dimers is discussed. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_20629.map.gz | 162.1 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-20629-v30.xmlemd-20629.xml | 15.8 KB 15.8 KB | Display Display | EMDB header |
| Images |  emd_20629.png emd_20629.png | 30.7 KB | ||
| Filedesc metadata | emd-20629.cif.gz | 6.6 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-20629ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20629 http://ftp.pdbj.org/pub/emdb/structures/EMD-20629ftp://ftp.pdbj.org/pub/emdb/structures/EMD-20629 | HTTPS FTP |
-Related structure data
| Related structure data |  6u3gMC  6u3eC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |









-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_20629.map.gz / Format: CCP4 / Size: 2 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Volume 2 for truncated dimeric Cytohesin-3 (Grp1; amino acids 14-399) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 6.5 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Truncated homodimer of Cytohesin-3 (Grp1, amino acids 14-399) wit...
| Entire | Name: Truncated homodimer of Cytohesin-3 (Grp1, amino acids 14-399) with Inositol 1,3,4,5-tetrakis phosphate (IP4) |
|---|---|
| Components |
|
-Supramolecule #1: Truncated homodimer of Cytohesin-3 (Grp1, amino acids 14-399) wit...
| Supramolecule | Name: Truncated homodimer of Cytohesin-3 (Grp1, amino acids 14-399) with Inositol 1,3,4,5-tetrakis phosphate (IP4) type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
-Macromolecule #1: Cytohesin-3
| Macromolecule | Name: Cytohesin-3 / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 46.501766 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MGHHHHHHGS PEDLSLEERE ELLDIRRRKK ELIDDIERLK YEIAEVMTEI DNLTSVEESK YTQRNAQIAM GRKKFNMDPK KGIQFLIEN DLLQSSPEDV AQFLYKGEGL NKTVIGDYLG ERDDFNIKVL QAFVELHEFA DLNLVQALRQ FLWSFRLPGE A QKIDRMME ...String: MGHHHHHHGS PEDLSLEERE ELLDIRRRKK ELIDDIERLK YEIAEVMTEI DNLTSVEESK YTQRNAQIAM GRKKFNMDPK KGIQFLIEN DLLQSSPEDV AQFLYKGEGL NKTVIGDYLG ERDDFNIKVL QAFVELHEFA DLNLVQALRQ FLWSFRLPGE A QKIDRMME AFASRYCLCN PGVFQSTDTC YVLSFAIIML NTSLHNHNVR DKPTAERFIT MNRGINEGGD LPEELLRNLY ES IKNEPFK IPEDDGNDLT YTFFNPDREG WLLKLGGRVK TWKRRWFILT DNCLYYFEYT TDKEPRGIIP LENLSIREVE DPR KPNCFE LYNPSHKGQV IKACKTEADG RVVEGNHVVY RISAPSPEEK EEWMKSIKAS ISRDPFYDML ATRKRRIANK K UniProtKB: Cytohesin-3 |
-Macromolecule #2: INOSITOL-(1,3,4,5)-TETRAKISPHOSPHATE
| Macromolecule | Name: INOSITOL-(1,3,4,5)-TETRAKISPHOSPHATE / type: ligand / ID: 2 / Number of copies: 2 / Formula: 4IP |
|---|---|
| Molecular weight | Theoretical: 500.075 Da |
| Chemical component information | 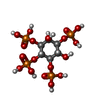 ChemComp-4IP: |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Buffer | pH: 8 Component:
Details: Peak fractions after gel filtration were immediately diluted, applied to freshly glow discharged grids, and stained with uranyl formate. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Staining | Type: NEGATIVE / Material: Uranyl Formate / Details: Stained with 0.75% (w/v) uranyl formate | |||||||||
| Grid | Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. | |||||||||
| Details | The sample is a uniform homodimer with significant conformational flexibility. |
- Electron microscopy
Electron microscopy
| Microscope | FEI/PHILIPS CM120T |
|---|---|
| Details | specimen holder: FISCHIONE INSTRUMENTS DUAL AXIS TOMOGRAPHY HOLDER |
| Image recording | Film or detector model: TVIPS TEMCAM-F224 (2k x 2k) / Digitization - Dimensions - Width: 2048 pixel / Digitization - Dimensions - Height: 2048 pixel / Number grids imaged: 1 / Number real images: 500 / Average electron dose: 30.0 e/Å2 |
| Electron beam | Acceleration voltage: 120 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: -3.2 µm / Nominal defocus min: -1.2 µm / Nominal magnification: 28000 |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: B / Chain - Residue range: 63-399 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Details | The model with the best correlation coefficient was selected by ADP_EM from a pool of 10000 models generated by RRT_SAMPLE using rigid bodies derived from 2R09 (autoinhibited core, amino acids 63-399) and a canonical antiparallel coiled coil (amino acids 18-53) built with CCBuilder. Flexible regions with reasonable geometry were modeled with MODELLER. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: Correlation coefficient |
| Output model | PDB-6u3g: |

