 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1912 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the Human Myeloma IgG2 Mat. | |||||||||
 Map data Map data | human IgG2 Mat density map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Human IgG2 / single particle 3D reconstruction / electron microscopy | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method | single particle reconstruction / Resolution: 17.8 Å | |||||||||
 Authors Authors | Ryazantsev S / Tischenko V / Nguyen C / Abramov V / Zaviyalov V | |||||||||
 Citation Citation | Journal: PLoS One / Year: 2013 Title: Three-dimensional structure of the human myeloma IgG2. Authors: Sergey Ryazantsev / Vladimir Tischenko / Christopher Nguyen / Vyacheslav Abramov / Vladimir Zav'yalov /  Abstract: Human immunoglobulin G, subclass 2 (hIgG2), plays an important role in immunity to bacterial pathogens and in numerous pathological conditions. However, there is a lack of information regarding the ...Human immunoglobulin G, subclass 2 (hIgG2), plays an important role in immunity to bacterial pathogens and in numerous pathological conditions. However, there is a lack of information regarding the three-dimensional (3D) structure of the hIgG2 molecule. We used electron microscopy (EM), differential scanning microcalorimetry (DSC) and fluorescence for structural analysis of the hIgG2. DSC and fluorescence indicated two types of interaction between CH1 domain of Fab (antigen-binding fragment/subunit) and CH2 domain of Fc (complement fixation fragment/subunit) simultaneously present in the sample: close interaction, which increases the thermostability of both, CH1 and CH2 domains, and weak (or no) interaction, which is typical for most IgGs but not hIgG2. Thermodynamics could not determine if both types of interactions are present within a single molecule. To address this question, EM was used. We employed a single-particle reconstruction and negative staining approach to reveal the three-dimensional structure of the hIgG2. A three-dimensional model of hIgG2 was created at 1.78 nm resolution. The hIgG2 is asymmetrical: one Fab subunit is in close proximity to the upper portion of the Fc subunit (CH2 domain) and the other Fab is distant from Fc. The plane of Fab subunits is nearly perpendicular to Fc. EM structure of the hIgG2 is in good agreement with thermodynamic data: a Fab distant from Fc should exhibit a lower melting temperature while a Fab interacting with Fc should exhibit a higher melting temperature. Both types of Fab subunits exist within one molecule resembling an A/B hIgG2 isoform introduced earlier on physicochemical level by Dillon et al. (2008). In such an arrangement, the access to the upper portion of Fc subunit is partially blocked by a Fab subunit. That might explain for instance why hIgG2 mildly activates complement and binds poorly to Fc receptors. Understanding of the three-dimensional structure of the hIgG2 should lead to better design of antibody-based therapeutics. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera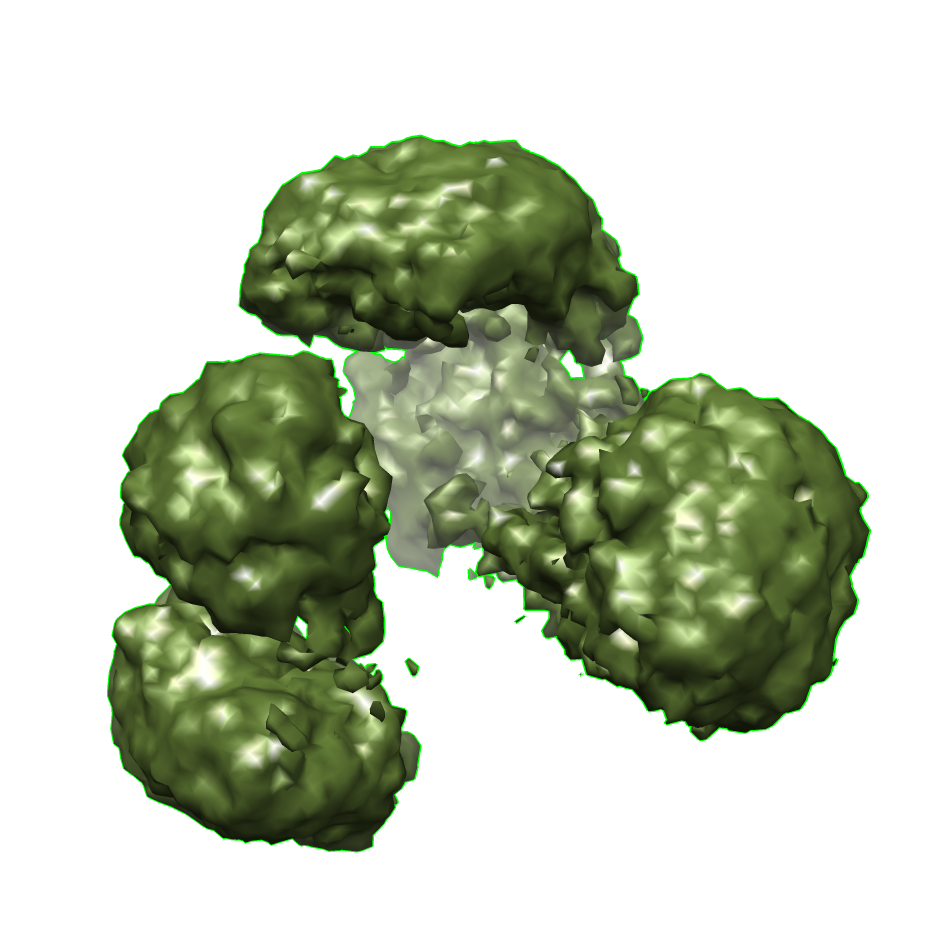
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1912.map.gz | 554.8 KB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1912-v30.xmlemd-1912.xml | 8.9 KB 8.9 KB | Display Display | EMDB header |
| Images |  emd-1912.png emd-1912.png | 361.1 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1912ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1912 http://ftp.pdbj.org/pub/emdb/structures/EMD-1912ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1912 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1912.map.gz / Format: CCP4 / Size: 2.9 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | human IgG2 Mat density map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 2.1 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Human myeloma immunoglobulin class G, subclass 2, MAT hIgG2 Mat
| Entire | Name: Human myeloma immunoglobulin class G, subclass 2, MAT hIgG2 Mat |
|---|---|
| Components |
|
-Supramolecule #1000: Human myeloma immunoglobulin class G, subclass 2, MAT hIgG2 Mat
| Supramolecule | Name: Human myeloma immunoglobulin class G, subclass 2, MAT hIgG2 Mat type: sample / ID: 1000 Details: More than 95 per cent purity. Stored at 4oC for 20 days before usage Oligomeric state: Monomer / Number unique components: 1 |
|---|---|
| Molecular weight | Experimental: 160 KDa / Theoretical: 160 KDa / Method: Gel-electrophoresis, sedimentation |
-Macromolecule #1: hIgG2 Mat
| Macromolecule | Name: hIgG2 Mat / type: protein_or_peptide / ID: 1 / Name.synonym: immunoglobulin G subclass two / Details: highly purified intact human myeloma IgG2 / Number of copies: 1 / Oligomeric state: Monomer / Recombinant expression: No |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) / Strain: hIgG2 Mat / synonym: Human / Tissue: Blood |
| Molecular weight | Experimental: 160 KDa / Theoretical: 160 KDa |
-Experimental details
-Structure determination
 Processing Processing | single particle reconstruction |
|---|---|
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.014 mg/mL |
|---|---|
| Buffer | pH: 7.8 / Details: 20 mM ammonium acetate, pH 7.8 |
| Grid | Details: 1.4 nm carbon film with carbon holey-film on top of 100 mesh copper hexagonal grid |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy
Electron microscopy
| Microscope | JEOL 1200EX |
|---|---|
| Alignment procedure | Legacy - Astigmatism: Corrected at x500,000 |
| Image recording | Category: CCD / Film or detector model: GENERIC FILM / Number real images: 2 / Details: 2.1 A per pixel / Bits/pixel: 16 |
| Tilt angle min | 0 |
| Tilt angle max | 0 |
| Electron beam | Acceleration voltage: 80 kV / Electron source: TUNGSTEN HAIRPIN |
| Electron optics | Calibrated magnification: 60000 / Illumination mode: OTHER / Imaging mode: BRIGHT FIELD / Cs: 1.4 mm / Nominal defocus max: 0.0 µm / Nominal defocus min: 0.0 µm / Nominal magnification: 60000 |
| Sample stage | Specimen holder: Normal / Specimen holder model: JEOL |
-Image processing
| Details | Images were taken at in-focus condition. No CTF correction was performed. |
|---|---|
| CTF correction | Details: No correction |
| Final reconstruction | Applied symmetry - Point group: C1 (asymmetric) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 17.8 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: EMAN / Number images used: 3000 |
| Final angle assignment | Details: Euler space was filled uniformly |
