Journal: mBio / Year: 2021 Title: SAMase of Bacteriophage T3 Inactivates Escherichia coli's Methionine -Adenosyltransferase by Forming Heteropolymers. Authors: Hadas Simon-Baram / Daniel Kleiner / Fannia Shmulevich / Raz Zarivach / Ran Zalk / Huayuan Tang / Feng Ding / Shimon Bershtein / Abstract: -Adenosylmethionine lyase (SAMase) of bacteriophage T3 degrades the intracellular SAM pools of the host Escherichia coli cells, thereby inactivating a crucial metabolite involved in a plethora of ...-Adenosylmethionine lyase (SAMase) of bacteriophage T3 degrades the intracellular SAM pools of the host Escherichia coli cells, thereby inactivating a crucial metabolite involved in a plethora of cellular functions, including DNA methylation. SAMase is the first viral protein expressed upon infection, and its activity prevents methylation of the T3 genome. Maintenance of the phage genome in a fully unmethylated state has a profound effect on the infection strategy. It allows T3 to shift from a lytic infection under normal growth conditions to a transient lysogenic infection under glucose starvation. Using single-particle cryoelectron microscopy (cryo-EM) and biochemical assays, we demonstrate that SAMase performs its function by not only degrading SAM but also by interacting with and efficiently inhibiting the host's methionine -adenosyltransferase (MAT), the enzyme that produces SAM. Specifically, SAMase triggers open-ended head-to-tail assembly of E. coli MAT into an unusual linear filamentous structure in which adjacent MAT tetramers are joined by two SAMase dimers. Molecular dynamics simulations together with normal mode analyses suggest that the entrapment of MAT tetramers within filaments leads to an allosteric inhibition of MAT activity due to a shift to low-frequency, high-amplitude active-site-deforming modes. The amplification of uncorrelated motions between active-site residues weakens MAT's substrate binding affinity, providing a possible explanation for the observed loss of function. We propose that the dual function of SAMase as an enzyme that degrades SAM and as an inhibitor of MAT activity has emerged to achieve an efficient depletion of the intracellular SAM pools. Self-assembly of enzymes into filamentous structures in response to specific metabolic cues has recently emerged as a widespread strategy of metabolic regulation. In many instances, filamentation of metabolic enzymes occurs in response to starvation and leads to functional inactivation. Here, we report that bacteriophage T3 modulates the metabolism of the host E. coli cells by recruiting a similar strategy: silencing a central metabolic enzyme by subjecting it to phage-mediated polymerization. This observation points to an intriguing possibility that virus-induced polymerization of the host metabolic enzymes is a common mechanism implemented by viruses to metabolically reprogram and subdue infected cells.
History
Deposition
Apr 27, 2021
-
Header (metadata) release
Jul 21, 2021
-
Map release
Jul 21, 2021
-
Update
Jul 2, 2025
-
Current status
Jul 2, 2025
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Keywords
Keywords Function and homology information
Function and homology information
 Escherichia virus T3 /
Escherichia virus T3 /  Authors
Authors Israel, 1 items
Israel, 1 items  Citation
Citation
 Structure visualization
Structure visualization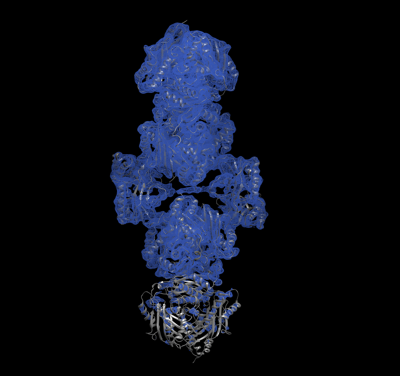
 Downloads & links
Downloads & links emd_12809.png
emd_12809.png http://ftp.pdbj.org/pub/emdb/structures/EMD-12809
http://ftp.pdbj.org/pub/emdb/structures/EMD-12809








 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
