 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-0603: Cryo-EM structure of the human TFIIH core complex: Multibody-refi... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-0603 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
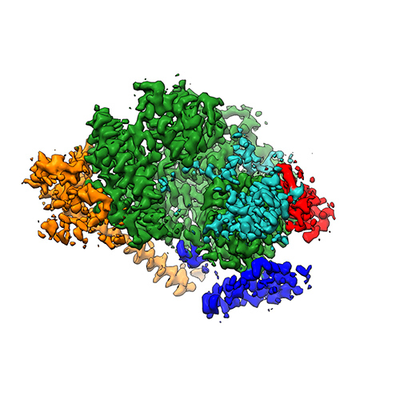
| Title | Cryo-EM structure of the human TFIIH core complex: Multibody-refined map, body 2 | |||||||||||||||
 Map data Map data | Multibody-refined cryo-EM map, body 2 (post-processed). | |||||||||||||||
 Sample Sample |
| |||||||||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.6 Å | |||||||||||||||
 Authors Authors | Greber BJ / Toso D / Fang J / Nogales E | |||||||||||||||
| Funding support |  United States, United States,  Switzerland, 4 items Switzerland, 4 items
| |||||||||||||||
 Citation Citation | Journal: Elife / Year: 2019 Title: The complete structure of the human TFIIH core complex. Authors: Basil J Greber / Daniel B Toso / Jie Fang / Eva Nogales / Abstract: Transcription factor IIH (TFIIH) is a heterodecameric protein complex critical for transcription initiation by RNA polymerase II and nucleotide excision DNA repair. The TFIIH core complex is ...Transcription factor IIH (TFIIH) is a heterodecameric protein complex critical for transcription initiation by RNA polymerase II and nucleotide excision DNA repair. The TFIIH core complex is sufficient for its repair functions and harbors the XPB and XPD DNA-dependent ATPase/helicase subunits, which are affected by human disease mutations. Transcription initiation additionally requires the CdK activating kinase subcomplex. Previous structural work has provided only partial insight into the architecture of TFIIH and its interactions within transcription pre-initiation complexes. Here, we present the complete structure of the human TFIIH core complex, determined by phase-plate cryo-electron microscopy at 3.7 Å resolution. The structure uncovers the molecular basis of TFIIH assembly, revealing how the recruitment of XPB by p52 depends on a pseudo-symmetric dimer of homologous domains in these two proteins. The structure also suggests a function for p62 in the regulation of XPD, and allows the mapping of previously unresolved human disease mutations. | |||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_0603.map.gz | 60 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-0603-v30.xmlemd-0603.xml | 19.9 KB 19.9 KB | Display Display | EMDB header |
| Images |  emd_0603.png emd_0603.png | 132.9 KB | ||
| Masks | emd_0603_msk_1.map | 64 MB | Mask map | |
| Others | emd_0603_half_map_1.map.gzemd_0603_half_map_2.map.gz | 40 MB 40 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-0603ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0603 http://ftp.pdbj.org/pub/emdb/structures/EMD-0603ftp://ftp.pdbj.org/pub/emdb/structures/EMD-0603 | HTTPS FTP |
-Related structure data
| Related structure data |  0452C  0587C  0588C  0589C  0602C  0604C  6nmiC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_0603.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Multibody-refined cryo-EM map, body 2 (post-processed). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.15 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_0603_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |






-Half map: Unfiltered half-map.
| File | emd_0603_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered half-map. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: Unfiltered half-map.
| File | emd_0603_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Unfiltered half-map. | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |







- Sample components
Sample components
-Entire : Transcription factor IIH (TFIIH)
| Entire | Name: Transcription factor IIH (TFIIH) |
|---|---|
| Components |
|
-Supramolecule #1: Transcription factor IIH (TFIIH)
| Supramolecule | Name: Transcription factor IIH (TFIIH) / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#8 |
|---|---|
| Source (natural) | Organism: Homo sapiens (human) |
| Molecular weight | Theoretical: 500 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.0049 mg/mL | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.9 Component:
| ||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV Details: A thin film of continuous carbon was floated onto Protochips C-flat CF-4/2 holey carbon grids and glow discharged or plasma cleaned before application of 4 uL of sample solution.. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE / Energy filter - Name: GIF Quantum LS / Energy filter - Slit width: 20 eV |
| Details | Residual beam tilt corrected in RELION 3. |
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: SUPER-RESOLUTION / Number grids imaged: 7 / Number real images: 21437 / Average exposure time: 8.25 sec. / Average electron dose: 50.0 e/Å2 Details: Images were collected as dose-fractionated movie frames (33 or 50 frames per exposure). |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 43478 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 0.7 µm / Nominal defocus min: 0.5 µm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Details | This map supported model re-building in O and COOT. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT |
