 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- SASDDN3: Bovine serum albumin mixture: averaged and individual data frames... -
+ Open data
Open data

- Basic information
Basic information
| Entry | 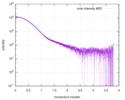 |
|---|---|
 Sample Sample | Bovine serum albumin mixture: averaged and individual data frames (subtracted and unsubtracted test sets)
|
| Function / homology |  Function and homology information Function and homology informationcellular response to calcium ion starvation / enterobactin binding / negative regulation of mitochondrial depolarization / toxic substance binding / fatty acid binding / cellular response to starvation / pyridoxal phosphate binding / protein-containing complex / DNA binding / extracellular space ...cellular response to calcium ion starvation / enterobactin binding / negative regulation of mitochondrial depolarization / toxic substance binding / fatty acid binding / cellular response to starvation / pyridoxal phosphate binding / protein-containing complex / DNA binding / extracellular space / extracellular region / metal ion binding / cytoplasm Similarity search - Function |
| Biological species |  |
 Citation Citation | Journal: Biophys J / Year: 2018 Title: Machine Learning Methods for X-Ray Scattering Data Analysis from Biomacromolecular Solutions. Authors: Daniel Franke / Cy M Jeffries / Dmitri I Svergun /  Abstract: Small-angle x-ray scattering (SAXS) of biological macromolecules in solutions is a widely employed method in structural biology. SAXS patterns include information about the overall shape and low- ...Small-angle x-ray scattering (SAXS) of biological macromolecules in solutions is a widely employed method in structural biology. SAXS patterns include information about the overall shape and low-resolution structure of dissolved particles. Here, we describe how to transform experimental SAXS patterns to feature vectors and how a simple k-nearest neighbor approach is able to retrieve information on overall particle shape and maximal diameter (D) as well as molecular mass directly from experimental scattering data. Based on this transformation, we develop a rapid multiclass shape-classification ranging from compact, extended, and flat categories to hollow and random-chain-like objects. This classification may be employed, e.g., as a decision block in automated data analysis pipelines. Further, we map protein structures from the Protein Data Bank into the classification space and, in a second step, use this mapping as a data source to obtain accurate estimates for the structural parameters (D, molecular mass) of the macromolecule under study based on the experimental scattering pattern alone, without inverse Fourier transform for D. All methods presented are implemented in a Fortran binary DATCLASS, part of the ATSAS data analysis suite, available on Linux, Mac, and Windows and free for academic use. |
 Contact author Contact author |
|
- Structure visualization
Structure visualization
- Downloads & links
Downloads & links
-Data source
| SASBDB page |  SASDDN3 SASDDN3 |
|---|
-Related structure data
| Related structure data | C: citing same article ( |
|---|---|
| Similar structure data |


-External links
| Related items in Molecule of the Month |
|---|




-Models
-Sample
| Sample | Name: Bovine serum albumin mixture: averaged and individual data frames (subtracted and unsubtracted test sets) Specimen concentration: 2.25 mg/ml |
|---|---|
| Buffer | Name: 50 mM HEPES / Concentration: 50.00 mM / pH: 7.5 |
| Entity #969 | Name: BSA / Type: protein / Description: Bovine serum albumin / Formula weight: 66.462 / Source: Bos taurus / References: UniProt: P02769 Sequence: DTHKSEIAHR FKDLGEEHFK GLVLIAFSQY LQQCPFDEHV KLVNELTEFA KTCVADESHA GCEKSLHTLF GDELCKVASL RETYGDMADC CEKQEPERNE CFLSHKDDSP DLPKLKPDPN TLCDEFKADE KKFWGKYLYE IARRHPYFYA PELLYYANKY NGVFQECCQA ...Sequence: DTHKSEIAHR FKDLGEEHFK GLVLIAFSQY LQQCPFDEHV KLVNELTEFA KTCVADESHA GCEKSLHTLF GDELCKVASL RETYGDMADC CEKQEPERNE CFLSHKDDSP DLPKLKPDPN TLCDEFKADE KKFWGKYLYE IARRHPYFYA PELLYYANKY NGVFQECCQA EDKGACLLPK IETMREKVLT SSARQRLRCA SIQKFGERAL KAWSVARLSQ KFPKAEFVEV TKLVTDLTKV HKECCHGDLL ECADDRADLA KYICDNQDTI SSKLKECCDK PLLEKSHCIA EVEKDAIPEN LPPLTADFAE DKDVCKNYQE AKDAFLGSFL YEYSRRHPEY AVSVLLRLAK EYEATLEECC AKDDPHACYS TVFDKLKHLV DEPQNLIKQN CDQFEKLGEY GFQNALIVRY TRKVPQVSTP TLVEVSRSLG KVGTRCCTKP ESERMPCTED YLSLILNRLC VLHEKTPVSE KVTKCCTESL VNRRPCFSAL TPDETYVPKA FDEKLFTFHA DICTLPDTEK QIKKQTALVE LLKHKPKATE EQLKTVMENF VAFVDKCCAA DDKEACFAVE GPKLVVSTQT ALA |
-Experimental information
| Beam | Instrument name: PETRA III EMBL P12 / City: Hamburg / 国: Germany / Type of source: X-ray synchrotron / Wavelength: 0.124 Å / Dist. spec. to detc.: 3.1 mm | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Detector | Name: Pilatus 2M | ||||||||||||||||||||||||||||||
| Scan | 
| ||||||||||||||||||||||||||||||
| Distance distribution function P(R) | 
| ||||||||||||||||||||||||||||||
| Result |  Comments: The data displayed in this entry represents the averaged 1D-SAXS profile obtained from 100 individual SAXS data frames. Included in the full entry zip-archive are each of the individual ...Comments: The data displayed in this entry represents the averaged 1D-SAXS profile obtained from 100 individual SAXS data frames. Included in the full entry zip-archive are each of the individual subtracted SAXS profiles as well as each individual sample and buffer frame (100 in total).
|
