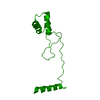
ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 8tua | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Full-length P-Rex1 in complex with inositol 1,3,4,5-tetrakisphosphate (IP4) | ||||||||||||||||||
 要素 要素 | Phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 1 protein | ||||||||||||||||||
 キーワード キーワード | SIGNALING PROTEIN / Rho guanine-nucleotide exchange factor / Dbl homology domain / pleckstrin homology domain / Phosphatidylinositol 3 / 4 / 5-trisphosphate binding | ||||||||||||||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報regulation of signaling / regulation of dendrite development / neutrophil activation / regulation of actin filament polymerization / regulation of small GTPase mediated signal transduction / NRAGE signals death through JNK / RHOB GTPase cycle / superoxide metabolic process / RHOC GTPase cycle / RHOJ GTPase cycle ...regulation of signaling / regulation of dendrite development / neutrophil activation / regulation of actin filament polymerization / regulation of small GTPase mediated signal transduction / NRAGE signals death through JNK / RHOB GTPase cycle / superoxide metabolic process / RHOC GTPase cycle / RHOJ GTPase cycle / RHOQ GTPase cycle / CDC42 GTPase cycle / T cell differentiation / RHOG GTPase cycle / RHOA GTPase cycle / RAC2 GTPase cycle / RAC3 GTPase cycle / positive regulation of substrate adhesion-dependent cell spreading / neutrophil chemotaxis / RAC1 GTPase cycle / actin filament polymerization / GTPase activator activity / guanyl-nucleotide exchange factor activity / dendritic shaft / phospholipid binding / G alpha (12/13) signalling events / growth cone / intracellular signal transduction / positive regulation of cell migration / G protein-coupled receptor signaling pathway / perinuclear region of cytoplasm / enzyme binding / plasma membrane / cytosol 類似検索 - 分子機能 | ||||||||||||||||||
| 生物種 |  Homo sapiens (ヒト) Homo sapiens (ヒト) | ||||||||||||||||||
| 手法 | 電子顕微鏡法 / 単粒子再構成法 / クライオ電子顕微鏡法 / 解像度: 4.1 Å | ||||||||||||||||||
 データ登録者 データ登録者 | Cash, J.N. / Tesmer, J.J.G. | ||||||||||||||||||
| 資金援助 |  米国, 5件 米国, 5件
| ||||||||||||||||||
 引用 引用 | ジャーナル: Elife / 年: 2024 タイトル: Full-length P-Rex1 in complex with inositol 1,3,4,5-tetrakisphosphate (IP4) 著者: Ravala, S.K. / Adame-Garcia, S.R. / Li, S. / Chen, C. / Cianfrocco, M.A. / Gutkind, J.S. / Cash, J.N. / Tesmer, J.J.G. #1: ジャーナル: Acta Crystallogr D Struct Biol / 年: 2019 タイトル: Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. 著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / ...著者: Dorothee Liebschner / Pavel V Afonine / Matthew L Baker / Gábor Bunkóczi / Vincent B Chen / Tristan I Croll / Bradley Hintze / Li Wei Hung / Swati Jain / Airlie J McCoy / Nigel W Moriarty / Robert D Oeffner / Billy K Poon / Michael G Prisant / Randy J Read / Jane S Richardson / David C Richardson / Massimo D Sammito / Oleg V Sobolev / Duncan H Stockwell / Thomas C Terwilliger / Alexandre G Urzhumtsev / Lizbeth L Videau / Christopher J Williams / Paul D Adams /   要旨: Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological ...Diffraction (X-ray, neutron and electron) and electron cryo-microscopy are powerful methods to determine three-dimensional macromolecular structures, which are required to understand biological processes and to develop new therapeutics against diseases. The overall structure-solution workflow is similar for these techniques, but nuances exist because the properties of the reduced experimental data are different. Software tools for structure determination should therefore be tailored for each method. Phenix is a comprehensive software package for macromolecular structure determination that handles data from any of these techniques. Tasks performed with Phenix include data-quality assessment, map improvement, model building, the validation/rebuilding/refinement cycle and deposition. Each tool caters to the type of experimental data. The design of Phenix emphasizes the automation of procedures, where possible, to minimize repetitive and time-consuming manual tasks, while default parameters are chosen to encourage best practice. A graphical user interface provides access to many command-line features of Phenix and streamlines the transition between programs, project tracking and re-running of previous tasks. #2: ジャーナル: Nat Methods / 年: 2019タイトル: Real-time cryo-electron microscopy data preprocessing with Warp. 著者: Dimitry Tegunov / Patrick Cramer /  要旨: The acquisition of cryo-electron microscopy (cryo-EM) data from biological specimens must be tightly coupled to data preprocessing to ensure the best data quality and microscope usage. Here we ...The acquisition of cryo-electron microscopy (cryo-EM) data from biological specimens must be tightly coupled to data preprocessing to ensure the best data quality and microscope usage. Here we describe Warp, a software that automates all preprocessing steps of cryo-EM data acquisition and enables real-time evaluation. Warp corrects micrographs for global and local motion, estimates the local defocus and monitors key parameters for each recorded micrograph or tomographic tilt series in real time. The software further includes deep-learning-based models for accurate particle picking and image denoising. The output from Warp can be fed into established programs for particle classification and 3D-map refinement. Our benchmarks show improvement in the nominal resolution, which went from 3.9 Å to 3.2 Å, of a published cryo-EM data set for influenza virus hemagglutinin. Warp is easy to install from http://github.com/cramerlab/warp and computationally inexpensive, and has an intuitive, streamlined user interface. #3: ジャーナル: Nat Methods / 年: 2020 タイトル: Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. 著者: Ali Punjani / Haowei Zhang / David J Fleet /  要旨: Cryogenic electron microscopy (cryo-EM) is widely used to study biological macromolecules that comprise regions with disorder, flexibility or partial occupancy. For example, membrane proteins are ...Cryogenic electron microscopy (cryo-EM) is widely used to study biological macromolecules that comprise regions with disorder, flexibility or partial occupancy. For example, membrane proteins are often kept in solution with detergent micelles and lipid nanodiscs that are locally disordered. Such spatial variability negatively impacts computational three-dimensional (3D) reconstruction with existing iterative refinement algorithms that assume rigidity. We introduce non-uniform refinement, an algorithm based on cross-validation optimization, which automatically regularizes 3D density maps during refinement to account for spatial variability. Unlike common shift-invariant regularizers, non-uniform refinement systematically removes noise from disordered regions, while retaining signal useful for aligning particle images, yielding dramatically improved resolution and 3D map quality in many cases. We obtain high-resolution reconstructions for multiple membrane proteins as small as 100 kDa, demonstrating increased effectiveness of cryo-EM for this class of targets critical in structural biology and drug discovery. Non-uniform refinement is implemented in the cryoSPARC software package. | ||||||||||||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 8tua.cif.gz | 548.4 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb8tua.ent.gz | 371 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 8tua.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 8tua_validation.pdf.gz | 1.3 MB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 8tua_full_validation.pdf.gz | 1.3 MB | 表示 | |
| XML形式データ | 8tua_validation.xml.gz | 54.3 KB | 表示 | |
| CIF形式データ | 8tua_validation.cif.gz | 80.2 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/tu/8tuaftp://data.pdbj.org/pub/pdb/validation_reports/tu/8tua | HTTPS FTP |
-関連構造データ
| 関連構造データ |  41621MC M: このデータのモデリングに利用したマップデータ C: 同じ文献を引用 ( |
|---|---|
| 類似構造データ | |
| 実験データセット #1 | データ参照: 10.6019/EMPIAR-11967 / データの種類: EMPIAR |
-リンク
 PDBj
PDBj




- 集合体
集合体
| 登録構造単位 | 
|
|---|---|
| 1 |
|
-要素
| #1: タンパク質 | 分子量: 183459.484 Da / 分子数: 1 / 由来タイプ: 組換発現 / 詳細: Inositol 1,3,4,5-tetrakisphosphate / 由来: (組換発現) Homo sapiens (ヒト) / 遺伝子: PREX1 / 細胞株 (発現宿主): 293 Freestyle / 発現宿主: Homo sapiens (ヒト) / 参照: UniProt: Q8TCU6 |
|---|---|
| #2: 化合物 | ChemComp-4IP / 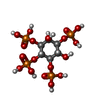  分子量: 500.075 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H16O18P4 / タイプ: SUBJECT OF INVESTIGATION 分子量: 500.075 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C6H16O18P4 / タイプ: SUBJECT OF INVESTIGATION |
| 研究の焦点であるリガンドがあるか | Y |
-実験情報
-実験
| 実験 | 手法: 電子顕微鏡法 |
|---|---|
| EM実験 | 試料の集合状態: PARTICLE / 3次元再構成法: 単粒子再構成法 |
- 試料調製
試料調製
| 構成要素 | 名称: Complex of full-length Phosphatidylinositol 3,4,5-trisphosphate (PIP3)-dependent Rac exchanger 1 (P-Rex1) bound to inositol 1,3,4,5-tetrakisphosphate (IP4) タイプ: COMPLEX / Entity ID: #1 / 由来: RECOMBINANT | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 分子量 | 値: 0.183 MDa / 実験値: NO | ||||||||||||||||||||
| 由来(天然) | 生物種: Homo sapiens (ヒト) | ||||||||||||||||||||
| 由来(組換発現) | 生物種: Homo sapiens (ヒト) / 株: HEK293 Freestyle | ||||||||||||||||||||
| 緩衝液 | pH: 8 | ||||||||||||||||||||
| 緩衝液成分 |
| ||||||||||||||||||||
| 試料 | 濃度: 0.56 mg/ml / 包埋: NO / シャドウイング: NO / 染色: NO / 凍結: YES | ||||||||||||||||||||
| 試料支持 | グリッドの材料: COPPER / グリッドのサイズ: 300 divisions/in. / グリッドのタイプ: Quantifoil R1.2/1.3 | ||||||||||||||||||||
| 急速凍結 | 装置: FEI VITROBOT MARK IV / 凍結剤: ETHANE / 湿度: 100 % / 凍結前の試料温度: 277 K |
- 電子顕微鏡撮影
電子顕微鏡撮影
| 実験機器 |  モデル: Titan Krios / 画像提供: FEI Company |
|---|---|
| 顕微鏡 | モデル: FEI TITAN KRIOS |
| 電子銃 | 電子線源:  FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM FIELD EMISSION GUN / 加速電圧: 300 kV / 照射モード: FLOOD BEAM |
| 電子レンズ | モード: BRIGHT FIELD / 倍率(公称値): 81000 X / 最大 デフォーカス(公称値): 2000 nm / 最小 デフォーカス(公称値): 200 nm / Cs: 2.7 mm |
| 試料ホルダ | 凍結剤: NITROGEN 試料ホルダーモデル: FEI TITAN KRIOS AUTOGRID HOLDER |
| 撮影 | 電子線照射量: 57.8 e/Å2 / フィルム・検出器のモデル: GATAN K3 (6k x 4k) |
- 解析
解析
| EMソフトウェア | 名称: PHENIX / バージョン: 1.21_5207 / カテゴリ: モデル精密化 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF補正 | タイプ: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||
| 粒子像の選択 | 選択した粒子像数: 2426612 詳細: 806,067 particles untilted and 1,620,545 particles tilted | ||||||||||||||||||||||||
| 対称性 | 点対称性: C1 (非対称) | ||||||||||||||||||||||||
| 3次元再構成 | 解像度: 4.1 Å / 解像度の算出法: FSC 0.143 CUT-OFF / 粒子像の数: 187734 / 対称性のタイプ: POINT | ||||||||||||||||||||||||
| 原子モデル構築 | プロトコル: RIGID BODY FIT / 空間: REAL | ||||||||||||||||||||||||
| 原子モデル構築 | 3D fitting-ID: 1 / Chain-ID: A / PDB chain-ID: A / Source name: PDB / タイプ: experimental model
| ||||||||||||||||||||||||
| 精密化 | 交差検証法: NONE 立体化学のターゲット値: GeoStd + Monomer Library + CDL v1.2 | ||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 103.39 Å2 | ||||||||||||||||||||||||
| 拘束条件 |
|