National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
P41GM136508
United States
Citation
Journal: Nat Methods / Year: 2022 Title: Ab initio phasing macromolecular structures using electron-counted MicroED data. Authors: Michael W Martynowycz / Max T B Clabbers / Johan Hattne / Tamir Gonen / Abstract: Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data that were collected on a direct electron detector in counting mode. Microcrystals ...Structures of two globular proteins were determined ab initio using microcrystal electron diffraction (MicroED) data that were collected on a direct electron detector in counting mode. Microcrystals were identified using a scanning electron microscope (SEM) and thinned with a focused ion beam (FIB) to produce crystalline lamellae of ideal thickness. Continuous-rotation data were collected using an ultra-low exposure rate to enable electron counting in diffraction. For the first sample, triclinic lysozyme extending to a resolution of 0.87 Å, an ideal helical fragment of only three alanine residues provided initial phases. These phases were improved using density modification, allowing the entire atomic structure to be built automatically. A similar approach was successful on a second macromolecular sample, proteinase K, which is much larger and diffracted to a resolution of 1.5 Å. These results demonstrate that macromolecules can be determined to sub-ångström resolution by MicroED and that ab initio phasing can be successfully applied to counting data.
ProteinaseK / Endopeptidase K / Tritirachium alkaline proteinase
Mass: 28930.783 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Parengyodontium album (fungus) / References: UniProt: P06873, peptidase K
Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Temperature (max): 90 K / Temperature (min): 77 K
Image recording
Average exposure time: 0.5 sec. / Electron dose: 0.001 e/Å2 / Film or detector model: FEI FALCON IV (4k x 4k) / Num. of diffraction images: 840 / Num. of grids imaged: 1 / Num. of real images: 1 Details: 0.15 degrees per second, 0.5 second readout, 30 to -30 degrees
Image scans
Sampling size: 28 µm / Width: 2048 / Height: 2048
EM diffraction
Camera length: 1738 mm / Tilt angle list: -30,30
EM diffraction shell
Resolution: 1.53→1.5 Å / Fourier space coverage: 89.3 % / Multiplicity: 8.9 / Num. of structure factors: 1758 / Phase residual: 30 °
EM diffraction stats
Details: Phases were determined by placing 4 ideal helix fragments. These were extended by chain tracing and density modifications. Fourier space coverage: 98.87 % / High resolution: 1.5 Å / Num. of intensities measured: 416133 / Num. of structure factors: 39303 / Phase error: 20 ° / Phase error rejection criteria: None / Rmerge: 0.277 / Rsym: 0.087
-
Processing
Software
Name: REFMAC / Version: 5.8.0267 / Classification: refinement / Contact author: Garib N. Murshudov / Contact author email: garib[at]mrc-lmb.cam.ac.uk / Date: 2020-24-08 Description: (un)restrained refinement or idealisation of macromolecular structures
EM software
ID
Name
Category
8
REFMAC
modelrefinement
12
AIMLESS
crystallographymerging
13
REFMAC
3Dreconstruction
Image processing
Details: Binned by 2.
EM 3D crystal entity
∠α: 90 ° / ∠β: 90 ° / ∠γ: 90 ° / A: 67.08 Å / B: 67.08 Å / C: 106.78 Å / Space group name: P43212 / Space group num: 96
CTF correction
Type: NONE
3D reconstruction
Resolution method: DIFFRACTION PATTERN/LAYERLINES / Symmetry type: 3D CRYSTAL
Atomic model building
B value: 14.137 / Protocol: AB INITIO MODEL / Space: RECIPROCAL / Target criteria: Maximum likelihood
Refinement
Resolution: 1.5→43.386 Å / Cor.coef. Fo:Fc: 0.969 / Cor.coef. Fo:Fc free: 0.95 / SU B: 4.529 / SU ML: 0.067 / Cross valid method: FREE R-VALUE / ESU R: 0.085 / ESU R Free: 0.078 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Selection details
Rfree
0.2046
2001
5.091 %
Random
Rwork
0.1495
37302
-
-
all
0.152
-
-
-
obs
-
39303
98.848 %
-
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Parengyodontium album (fungus)
Parengyodontium album (fungus) Authors
Authors United States, 2items
United States, 2items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads


 PDBj
PDBj

 Assembly
Assembly
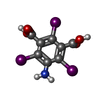
 Mass: 558.835 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H4I3NO4
Mass: 558.835 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H4I3NO4
 Mass: 40.078 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: Ca Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation
 FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM
FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM Processing
Processing