 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-11nf: Azotobacter vinelandii MoFeP (C2 symmetry) determined using the S... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 11nf | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Azotobacter vinelandii MoFeP (C2 symmetry) determined using the SPT Labtech chameleon in the presence of 1x SurfACT | |||||||||||||||||||||||||||||||||
 Components Components | (Nitrogenase molybdenum-iron protein ...) x 2 | |||||||||||||||||||||||||||||||||
 Keywords Keywords | OXIDOREDUCTASE / Nitrogenase / FeMoCo / nitrogen / P-cluster / METAL BINDING PROTEIN | |||||||||||||||||||||||||||||||||
| Function / homology |  Function and homology information Function and homology informationnitrogen fixation / molybdenum-iron nitrogenase complex / nitrogenase / nitrogenase activity / iron-sulfur cluster binding / ATP binding / metal ion binding Similarity search - Function | |||||||||||||||||||||||||||||||||
| Biological species |  Azotobacter vinelandii (bacteria) Azotobacter vinelandii (bacteria) | |||||||||||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.34 Å | |||||||||||||||||||||||||||||||||
 Authors Authors | Enos, S.E. / Cook, B.D. / Rahmani, H. / Narehood, S.M. / Li, Y. / Kuschnerus, I.C. / Redford, H.T. / Dukakis, P. / Ji, D. / Bachochin, M.J. ...Enos, S.E. / Cook, B.D. / Rahmani, H. / Narehood, S.M. / Li, Y. / Kuschnerus, I.C. / Redford, H.T. / Dukakis, P. / Ji, D. / Bachochin, M.J. / Grotjahn, D.A. / Herzik, M.A. | |||||||||||||||||||||||||||||||||
| Funding support |  United States, 5items United States, 5items
| |||||||||||||||||||||||||||||||||
 Citation Citation | Journal: bioRxiv / Year: 2026 Title: A Surfactant Cocktail Overcomes Air-Water Interface Artifacts in Single-Particle CryoEM. Authors: Suzanne E Enos / Brian D Cook / Hamidreza Rahmani / Sarah M Narehood / Yizhou Li / Inga C Kuschnerus / Trevor H Redford / Peter Dukakis / Daniel Ji / Maxwell J Bachochin / Danielle A Grotjahn / Mark A Herzik / Abstract: Single-particle cryogenic electron microscopy (cryoEM) is a widely used technique for structure determination of biomacromolecules to near-atomic resolution. Random distributions of these molecules ...Single-particle cryogenic electron microscopy (cryoEM) is a widely used technique for structure determination of biomacromolecules to near-atomic resolution. Random distributions of these molecules in vitrified ice are necessary to accumulate enough two-dimensional views to generate a complete three-dimensional (3-D) reconstruction. However, interactions between the sample and the air-water interface (AWI) that occur during vitrification often bias the views of the sample, a phenomenon termed preferred orientation, limiting our ability to obtain 3-D reconstructions. Surfactants are often used as sample additives to prevent AWI-induced deterioration, but no general strategy exists for surfactant choice, requiring laborious screening for each sample. To circumvent these issues, we developed SurfACT, a cocktail of diverse surfactants with distinct physicochemical properties that limits AWI-dependent sample denaturation and orientation bias, while mitigating individual surfactant-specific drawbacks. Here we demonstrate SurfACT's effectiveness with four proteins plagued by AWI-induced issues, including two species of hemagglutinin (HA), molybdenum-iron protein (MoFeP) from the nitrogenase enzyme, and aldolase. All four samples show drastically improved viewing distribution and map completeness when SurfACT is applied. Cryogenic electron tomography demonstrates that SurfACT redistributes particles from the AWI into the bulk ice, driving signal recovery and inhibiting denaturation. This versatile sample additive minimizes sample-specific screening and expands the capabilities and range of suitable samples for cryoEM. | |||||||||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 11nf.cif.gz | 447.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb11nf.ent.gz | 355.7 KB | Display | PDB format |
| PDBx/mmJSON format | 11nf.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/1n/11nfftp://data.pdbj.org/pub/pdb/validation_reports/1n/11nf | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  75859MC  11msC  11mtC  11muC  11mvC  11mxC  11mzC  11naC  11nbC  11ncC  11ndC  11neC  11nhC  11niC  11njC  11nkC  11nlC  11nmC  11nnC  11noC  11npC  11nrC  11ntC  11nuC  11nwC  11nxC  11obC  11ocC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data |







































-Links
 PDBj
PDBj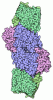
- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
-Nitrogenase molybdenum-iron protein ... , 2 types, 4 molecules ACBD
| #1: Protein | Mass: 55363.043 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Azotobacter vinelandii (bacteria) / References: UniProt: P07328, nitrogenase#2: Protein | Mass: 59535.879 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Azotobacter vinelandii (bacteria) / References: UniProt: P07329, nitrogenase |
|---|
-Non-polymers , 5 types, 1851 molecules 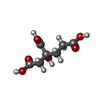
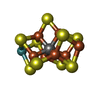

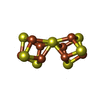

| #3: Chemical |  Mass: 206.150 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H10O7 / Feature type: SUBJECT OF INVESTIGATION Mass: 206.150 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C7H10O7 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |  Mass: 787.451 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CFe7MoS9 / Feature type: SUBJECT OF INVESTIGATION Mass: 787.451 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CFe7MoS9 / Feature type: SUBJECT OF INVESTIGATION#5: Chemical |  Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION Mass: 55.845 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe / Feature type: SUBJECT OF INVESTIGATION#6: Chemical |  Mass: 671.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe8S7 / Feature type: SUBJECT OF INVESTIGATION Mass: 671.215 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Fe8S7 / Feature type: SUBJECT OF INVESTIGATION#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1843 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | N |
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: Azotobacter vinelandii MoFeP (C2 symmetry) determined using the SPT Labtech chameleon in the presence of 1x SurfACT Type: COMPLEX / Entity ID: #1-#2 / Source: NATURAL | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular weight | Value: 0.230 MDa / Experimental value: NO | |||||||||||||||||||||||||||||||||||
| Source (natural) | Organism: Azotobacter vinelandii (bacteria) | |||||||||||||||||||||||||||||||||||
| Buffer solution | pH: 8 | |||||||||||||||||||||||||||||||||||
| Buffer component |
| |||||||||||||||||||||||||||||||||||
| Specimen | Conc.: 3.6 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES | |||||||||||||||||||||||||||||||||||
| Specimen support | Grid material: COPPER / Grid type: Quantifoil Active R1.2/0.8 | |||||||||||||||||||||||||||||||||||
| Vitrification | Instrument: SPT LABTECH CHAMELEON / Cryogen name: ETHANE / Humidity: 75 % / Chamber temperature: 298.15 K / Details: Samples were frozen with the SPT Labtech chameleon |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Nominal magnification: 165000 X / Nominal defocus max: 2500 nm / Nominal defocus min: 1000 nm / Cs: 2.7 mm / Alignment procedure: COMA FREE |
| Specimen holder | Cryogen: NITROGEN / Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER |
| Image recording | Average exposure time: 5 sec. / Electron dose: 65 e/Å2 / Film or detector model: TFS FALCON 4i (4k x 4k) / Num. of grids imaged: 1 / Num. of real images: 1080 |
| EM imaging optics | Energyfilter name: TFS Selectris X / Energyfilter slit width: 10 eV |
| Image scans | Width: 4096 / Height: 4096 |
- Processing
Processing
| EM software |
| ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTF correction | Type: PHASE FLIPPING AND AMPLITUDE CORRECTION | ||||||||||||||||||||||||||||||||
| Symmetry | Point symmetry: C2 (2 fold cyclic) | ||||||||||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.34 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 23561 / Algorithm: FOURIER SPACE / Symmetry type: POINT | ||||||||||||||||||||||||||||||||
| Atomic model building | B value: 49.9 / Protocol: RIGID BODY FIT / Space: REAL | ||||||||||||||||||||||||||||||||
| Atomic model building | PDB-ID: 7UT7 Accession code: 7UT7 / Source name: PDB / Type: experimental model | ||||||||||||||||||||||||||||||||
| Refinement | Highest resolution: 2.34 Å Stereochemistry target values: REAL-SPACE (WEIGHTED MAP SUM AT ATOM CENTERS) | ||||||||||||||||||||||||||||||||
| Refine LS restraints |
|

