 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-4789 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | The pore structure of Clostridium perfringens epsilon toxin | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | enterotoxaemia / beta-PFT / epsilon toxin / CryoEM / TOXIN | |||||||||
| Function / homology | symbiont-mediated pore formation in host plasma membrane / Epsilon toxin / Aerolysin-like toxin / Clostridium epsilon toxin ETX/Bacillus mosquitocidal toxin MTX2 / toxin activity / Epsilon-toxin type B Function and homology information Function and homology information | |||||||||
| Biological species |  Clostridium perfringens B (bacteria) Clostridium perfringens B (bacteria) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 3.2 Å | |||||||||
 Authors Authors | Savva CG / Clark AR | |||||||||
| Funding support |  United Kingdom, 2 items United Kingdom, 2 items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2019 Title: The pore structure of Clostridium perfringens epsilon toxin. Authors: Christos G Savva / Alice R Clark / Claire E Naylor / Michel R Popoff / David S Moss / Ajit K Basak / Richard W Titball / Monika Bokori-Brown /  Abstract: Epsilon toxin (Etx), a potent pore forming toxin (PFT) produced by Clostridium perfringens, is responsible for the pathogenesis of enterotoxaemia of ruminants and has been suggested to play a role in ...Epsilon toxin (Etx), a potent pore forming toxin (PFT) produced by Clostridium perfringens, is responsible for the pathogenesis of enterotoxaemia of ruminants and has been suggested to play a role in multiple sclerosis in humans. Etx is a member of the aerolysin family of β-PFTs (aβ-PFTs). While the Etx soluble monomer structure was solved in 2004, Etx pore structure has remained elusive due to the difficulty of isolating the pore complex. Here we show the cryo-electron microscopy structure of Etx pore assembled on the membrane of susceptible cells. The pore structure explains important mutant phenotypes and suggests that the double β-barrel, a common feature of the aβ-PFTs, may be an important structural element in driving efficient pore formation. These insights provide the framework for the development of novel therapeutics to prevent human and animal infections, and are relevant for nano-biotechnology applications. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_4789.map.gz | 4.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-4789-v30.xmlemd-4789.xml | 17.8 KB 17.8 KB | Display Display | EMDB header |
| Images | 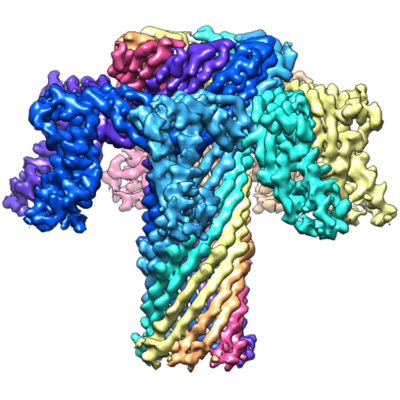 emd_4789.png emd_4789.png | 197.5 KB | ||
| Filedesc metadata | emd-4789.cif.gz | 6.1 KB | ||
| Others | emd_4789_half_map_1.map.gzemd_4789_half_map_2.map.gz | 31.3 MB 31.3 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-4789ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4789 http://ftp.pdbj.org/pub/emdb/structures/EMD-4789ftp://ftp.pdbj.org/pub/emdb/structures/EMD-4789 | HTTPS FTP |
-Related structure data
| Related structure data |  6rb9MC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_4789.map.gz / Format: CCP4 / Size: 40.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.07 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
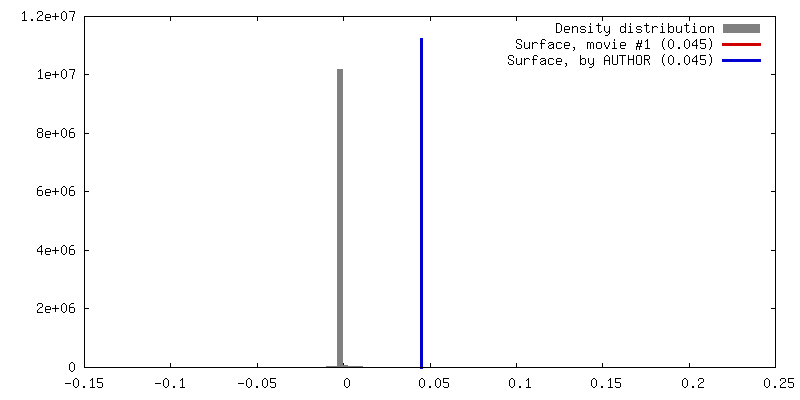
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: #1
| File | emd_4789_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |







-Half map: #2
| File | emd_4789_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Epsilon toxin
| Entire | Name: Epsilon toxin |
|---|---|
| Components |
|
-Supramolecule #1: Epsilon toxin
| Supramolecule | Name: Epsilon toxin / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Clostridium perfringens B (bacteria) |
| Molecular weight | Theoretical: 225.4 KDa |
-Macromolecule #1: Epsilon-toxin type B
| Macromolecule | Name: Epsilon-toxin type B / type: protein_or_peptide / ID: 1 / Number of copies: 7 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Clostridium perfringens B (bacteria) |
| Molecular weight | Theoretical: 32.234695 KDa |
| Recombinant expression | Organism: |
| Sequence | String: MSYYHHHHHH DYDIPTTENL YFQGAMASYD NVDTLIEKGR YNTKYNYLKR MEKYYPNAMA YFDKVTINPQ GNDFYINNPK VELDGEPSM NYLEDVYVGK ALLTNDTQQE QKLKSQSFTC KNTDTVTATT THTVGTSIQA TAKFTVPFNE TGVSLTTSYS F ANTNTNTN ...String: MSYYHHHHHH DYDIPTTENL YFQGAMASYD NVDTLIEKGR YNTKYNYLKR MEKYYPNAMA YFDKVTINPQ GNDFYINNPK VELDGEPSM NYLEDVYVGK ALLTNDTQQE QKLKSQSFTC KNTDTVTATT THTVGTSIQA TAKFTVPFNE TGVSLTTSYS F ANTNTNTN SKEITHNVPS QDILVPANTT VEVIAYLKKV NVKGNVKLVG QVSGSEWGEI PSYLAFPRDG YKFSLSDTVN KS DLNEDGT ININGKGNYS AVMGDELIVK VRNLNTNNVQ EYVIPVDKK UniProtKB: Epsilon-toxin type B |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.013 mg/mL |
|---|---|
| Buffer | pH: 7.1 Details: DPBS containing 20 mM imidazole and 0.02% (w/v) DDM |
| Grid | Model: Quantifoil R1.2/1.3 / Material: GOLD / Mesh: 300 / Support film - Material: GRAPHENE OXIDE / Support film - topology: CONTINUOUS / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 60 sec. / Pretreatment - Atmosphere: AIR / Details: 40 mA |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: HOMEMADE PLUNGER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Phase plate: VOLTA PHASE PLATE Chromatic aberration corrector: Volta phase plate was used. Just prior to data collection, the phase plate was inserted and the un-scattered beam was made parallel by observing the Ronchigram in the ...Chromatic aberration corrector: Volta phase plate was used. Just prior to data collection, the phase plate was inserted and the un-scattered beam was made parallel by observing the Ronchigram in the back focal plane of the objective |
| Details | AutoCTF |
| Image recording | Film or detector model: FEI FALCON III (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 835 / Average exposure time: 60.0 sec. / Average electron dose: 32.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 0.8 µm / Nominal defocus min: 0.3 µm / Nominal magnification: 75000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Details | A model was built into the 3.2 A map by initially docking the receptor-binding domain of the wild type Etx crystal structure (PDB: 1UYJ) and then extending this, building ab initio using Coot. |
|---|---|
| Refinement | Space: REAL / Protocol: OTHER |
| Output model | PDB-6rb9: |
