 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-1983: Symmetrized cryo-EM reconstruction of E. coli DegQ 12-mer in comp... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-1983 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Symmetrized cryo-EM reconstruction of E. coli DegQ 12-mer in complex with a binding peptide | |||||||||
 Map data Map data | Symmetrized reconstruction of Escherichia coli DegQ 12-mer in complex with a binding peptide | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Chaperone / Protease | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidase Do / protein quality control for misfolded or incompletely synthesized proteins / proteolysis involved in protein catabolic process / peptidase activity / periplasmic space / serine-type endopeptidase activity / proteolysis / identical protein binding Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 7.5 Å | |||||||||
 Authors Authors | Malet H / Canellas F / Sawa J / Yan J / Thalassinos K / Ehrmann M / Clausen T / Saibil HR | |||||||||
 Citation Citation | Journal: Nat Struct Mol Biol / Year: 2012 Title: Newly folded substrates inside the molecular cage of the HtrA chaperone DegQ. Authors: Hélène Malet / Flavia Canellas / Justyna Sawa / Jun Yan / Konstantinos Thalassinos / Michael Ehrmann / Tim Clausen / Helen R Saibil /  Abstract: The HtrA protein family combines chaperone and protease activities and is essential for protein quality control in many organisms. Whereas the mechanisms underlying the proteolytic function of HtrA ...The HtrA protein family combines chaperone and protease activities and is essential for protein quality control in many organisms. Whereas the mechanisms underlying the proteolytic function of HtrA proteins are well characterized, their chaperone activity remains poorly understood. Here we describe cryo-EM structures of Escherichia coli DegQ in its 12- and 24-mer states in complex with model substrates, providing a structural model of HtrA chaperone action. Up to six lysozyme substrates bind inside the DegQ 12-mer cage and are visualized in a close-to-native state. An asymmetric reconstruction reveals the binding of a well-ordered lysozyme to four DegQ protomers. DegQ PDZ domains are located adjacent to substrate density and their presence is required for chaperone activity. The substrate-interacting regions appear conserved in 12- and 24-mer cages, suggesting a common mechanism of chaperone function. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_1983.map.gz | 9.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-1983-v30.xmlemd-1983.xml | 13.9 KB 13.9 KB | Display Display | EMDB header |
| Images | 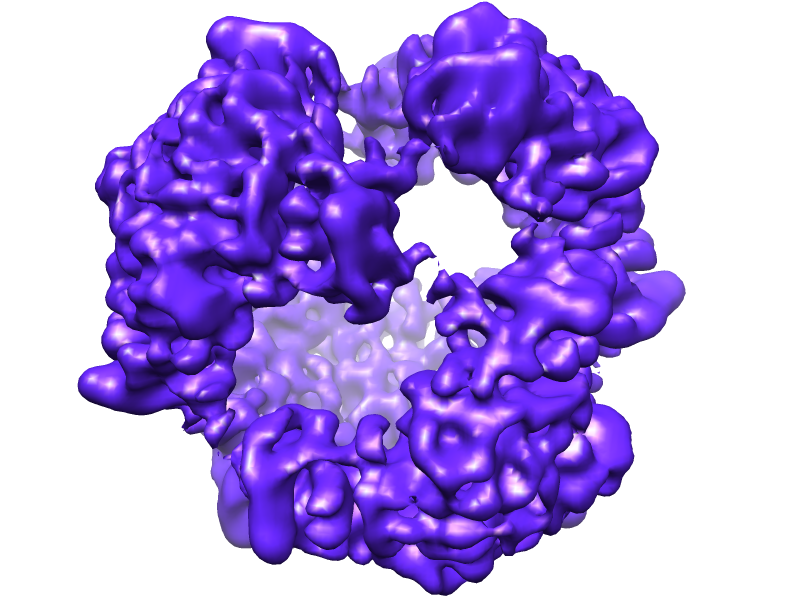 1983.png 1983.png | 354.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-1983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1983 http://ftp.pdbj.org/pub/emdb/structures/EMD-1983ftp://ftp.pdbj.org/pub/emdb/structures/EMD-1983 | HTTPS FTP |
-Validation report
| Summary document | emd_1983_validation.pdf.gz | 214.3 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_1983_full_validation.pdf.gz | 213.4 KB | Display | |
| Data in XML | emd_1983_validation.xml.gz | 6.4 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1983ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-1983 | HTTPS FTP |
-Related structure data
| Related structure data |  4a8cMC  1981C  1982C  1984C  4a8aC  4a8bC  4a8dC  4a9gC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |





-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_1983.map.gz / Format: CCP4 / Size: 62.5 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Symmetrized reconstruction of Escherichia coli DegQ 12-mer in complex with a binding peptide | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.4 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Escherichia coli DegQ 12-mer in complex with a binding peptide
| Entire | Name: Escherichia coli DegQ 12-mer in complex with a binding peptide |
|---|---|
| Components |
|
-Supramolecule #1000: Escherichia coli DegQ 12-mer in complex with a binding peptide
| Supramolecule | Name: Escherichia coli DegQ 12-mer in complex with a binding peptide type: sample / ID: 1000 / Details: - / Oligomeric state: Peptide monomer bound to one DegQ 12-mer / Number unique components: 2 |
|---|---|
| Molecular weight | Experimental: 540 KDa / Theoretical: 540 KDa / Method: Size exclusion chromatography |
-Macromolecule #1: DegQ
| Macromolecule | Name: DegQ / type: protein_or_peptide / ID: 1 / Name.synonym: DegQ / Number of copies: 12 / Oligomeric state: Dodecamer / Recombinant expression: Yes |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Experimental: 45 KDa / Theoretical: 45 KDa |
| Recombinant expression | Organism: |
-Macromolecule #2: peptide
| Macromolecule | Name: peptide / type: ligand / ID: 2 / Name.synonym: peptide / Details: Peptide sequence SPMFKGVLDMMYGGMRGYQV / Oligomeric state: Monomer / Recombinant expression: No |
|---|---|
| Source (natural) | Organism: synthetic construct (others) |
| Molecular weight | Experimental: 2.2 KDa / Theoretical: 2.2 KDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.2 mg/mL |
|---|---|
| Buffer | pH: 7.5 / Details: 20 mM HEPES/NaOH, 150 mM NaCl. |
| Grid | Details: C-flat grids (CF-2/2-4C-100 Protochips) |
| Vitrification | Cryogen name: ETHANE / Instrument: HOMEMADE PLUNGER / Details: Vitrification instrument: Manual plunger / Method: Blot for 2 seconds before plunging |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI F20 |
|---|---|
| Temperature | Min: 90 K / Max: 92 K / Average: 91 K |
| Alignment procedure | Legacy - Astigmatism: objective lens astigmatism was corrected at 150,000 times magnification Legacy - Electron beam tilt params: 0 |
| Details | Low dose mode |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: ZEISS SCAI / Digitization - Sampling interval: 7 µm / Number real images: 110 / Average electron dose: 15 e/Å2 / Bits/pixel: 8 |
| Tilt angle min | 0 |
| Tilt angle max | 0 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 50000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 50000 |
| Sample stage | Specimen holder: Single tilt cryo / Specimen holder model: GATAN LIQUID NITROGEN |
| Experimental equipment |  Model: Tecnai F20 / Image courtesy: FEI Company |
-Image processing
| CTF correction | Details: Phase flipping, full CTF correction |
|---|---|
| Final reconstruction | Applied symmetry - Point group: T (tetrahedral) / Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 7.5 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: IMAGIC-5, SPIDER / Number images used: 29432 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: Chimera, Flex-EM |
| Details | PDBEntryID_givenInChain. Protocol: Rigid body and flexible fitting. Protease and PDZ1 trimers extracted from PDB entry 3STJ. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Cross-correlation, energy |
| Output model | PDB-4a8c: |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - Chain ID: B |
|---|---|
| Software | Name: Chimera, Flex-EM |
| Details | PDBEntryID_givenInChain. Protocol: Rigid body and flexible fitting. Protease and PDZ1 trimers extracted from pdb entry 3STJ. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Cross-correlation, energy |
| Output model | PDB-4a8c: |
-Atomic model buiding 3
| Initial model | PDB ID: Chain - Chain ID: C |
|---|---|
| Software | Name: Chimera, Flex-EM |
| Details | PDBEntryID_givenInChain. Protocol: Rigid body and flexible fitting. Protease and PDZ1 trimers extracted from pdb entry 3STJ. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Cross-correlation, energy |
| Output model | PDB-4a8c: |
-Atomic model buiding 4
| Initial model | PDB ID: Chain - Chain ID: A |
|---|---|
| Software | Name: MODELLER, Chimera, Flex-EM |
| Details | PDBEntryID_givenInChain. Protocol: Rigid body and flexible fitting. PDZ2 domain (residues 377-466) modelled with MODELLER from E. coli DegP pdb entry 3CS0. |
| Refinement | Space: REAL / Protocol: FLEXIBLE FIT / Target criteria: Cross-correlation, energy |
| Output model | PDB-4a8c: |

