Journal: Gut Microbes / Year: 2020 Title: Functional analysis and cryo-electron microscopy of serine protease HtrA. Authors: Urszula Zarzecka / Alessandro Grinzato / Eaazhisai Kandiah / Dominik Cysewski / Paola Berto / Joanna Skorko-Glonek / Giuseppe Zanotti / Steffen Backert / Abstract: is a predominant zoonotic pathogen causing gastroenteritis and other diseases in humans. An important bacterial virulence factor is the secreted serine protease HtrA (HtrA ), which targets tight ... is a predominant zoonotic pathogen causing gastroenteritis and other diseases in humans. An important bacterial virulence factor is the secreted serine protease HtrA (HtrA ), which targets tight and adherens junctional proteins in the gut epithelium. Here we have investigated the function and structure of HtrA using biochemical assays and cryo-electron microscopy. Mass spectrometry analysis identified differences and similarities in the cleavage site specificity for HtrA by comparison to the HtrA counterparts from and . We defined the architecture of HtrA at 5.8 Å resolution as a dodecamer, built of four trimers. The contacts between the trimers are quite loose, a fact that explains the flexibility and mobility of the dodecameric assembly. This flexibility has also been studied through molecular dynamics simulation, which revealed opening of the dodecamer to expose the proteolytically active site of the protease. Moreover, we examined the rearrangements at the level of oligomerization in the presence or absence of substrate using size exclusion chromatography, which revealed hexamers, dodecamers and larger oligomeric forms, as well as remarkable stability of higher oligomeric forms (> 12-mers) compared to previously tested homologs from other bacteria. Extremely dynamic decay of the higher oligomeric forms into lower forms was observed after full cleavage of the substrate by the proteolytically active variant of HtrA . Together, this is the first report on the in-depth functional and structural analysis of HtrA , which may allow the construction of therapeutically relevant HtrA inhibitors in the near future.
History
Deposition
May 5, 2020
-
Header (metadata) release
Sep 30, 2020
-
Map release
Sep 30, 2020
-
Update
May 26, 2021
-
Current status
May 26, 2021
Processing site: PDBe / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information
 Campylobacter jejuni (Campylobacter)
Campylobacter jejuni (Campylobacter) Authors
Authors Citation
Citation



 Structure visualization
Structure visualization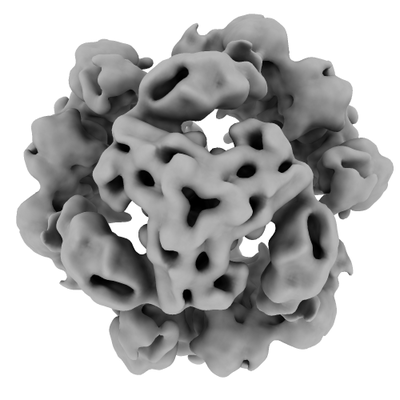
 Downloads & links
Downloads & links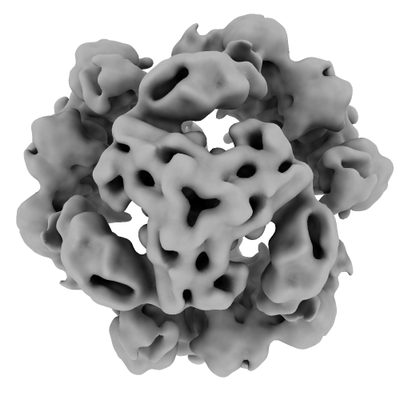 emd_11003.png
emd_11003.png http://ftp.pdbj.org/pub/emdb/structures/EMD-11003
http://ftp.pdbj.org/pub/emdb/structures/EMD-11003






 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)








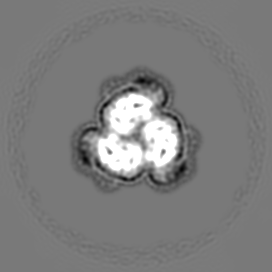












 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN