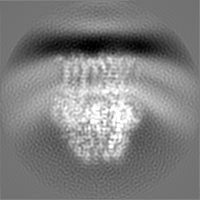
- EMDB-22050: 3.9 Angstrom reconstruction of E.coli AcrB embedded in the liposome -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: EMDB / ID: EMD-22050
Title
3.9 Angstrom reconstruction of E.coli AcrB embedded in the liposome
Map data
3D reconstruction of AcrB after Density Modification
Sample
Complex: E.coli AcrB
Function / homology
Function and homology information
alkane transmembrane transporter activity / alkane transport / enterobactin transport / enterobactin transmembrane transporter activity / xenobiotic detoxification by transmembrane export across the cell outer membrane / periplasmic side of plasma membrane / efflux pump complex / Iron assimilation using enterobactin / bile acid transmembrane transporter activity / Antimicrobial resistance ...alkane transmembrane transporter activity / alkane transport / enterobactin transport / enterobactin transmembrane transporter activity / xenobiotic detoxification by transmembrane export across the cell outer membrane / periplasmic side of plasma membrane / efflux pump complex / Iron assimilation using enterobactin / bile acid transmembrane transporter activity / Antimicrobial resistance / bile acid and bile salt transport / Secretion of toxins / efflux transmembrane transporter activity / xenobiotic transmembrane transporter activity / fatty acid transport / xenobiotic transport / response to toxic substance / response to xenobiotic stimulus / response to antibiotic / membrane / identical protein binding / plasma membrane Similarity search - Function
Journal: Proc Natl Acad Sci U S A / Year: 2020 Title: Cryo-EM analysis of a membrane protein embedded in the liposome. Authors: Xia Yao / Xiao Fan / Nieng Yan / Abstract: Membrane proteins (MPs) used to be the most difficult targets for structural biology when X-ray crystallography was the mainstream approach. With the resolution revolution of single-particle electron ...Membrane proteins (MPs) used to be the most difficult targets for structural biology when X-ray crystallography was the mainstream approach. With the resolution revolution of single-particle electron cryo-microscopy (cryo-EM), rapid progress has been made for structural elucidation of isolated MPs. The next challenge is to preserve the electrochemical gradients and membrane curvature for a comprehensive structural elucidation of MPs that rely on these chemical and physical properties for their biological functions. Toward this goal, here we present a convenient workflow for cryo-EM structural analysis of MPs embedded in liposomes, using the well-characterized AcrB as a prototype. Combining optimized proteoliposome isolation, cryo-sample preparation on graphene grids, and an efficient particle selection strategy, the three-dimensional (3D) reconstruction of AcrB embedded in liposomes was obtained at 3.9 Å resolution. The conformation of the homotrimeric AcrB remains the same when the surrounding membranes display different curvatures. Our approach, which can be widely applied to cryo-EM analysis of MPs with distinctive soluble domains, lays out the foundation for cryo-EM analysis of integral or peripheral MPs whose functions are affected by transmembrane electrochemical gradients or/and membrane curvatures.
History
Deposition
May 25, 2020
-
Header (metadata) release
Jul 15, 2020
-
Map release
Jul 15, 2020
-
Update
Aug 19, 2020
-
Current status
Aug 19, 2020
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
EMPIAR-10426 (Title: 3.9 Angstrom reconstruction of E.coli AcrB embedded in the liposome Data size: 9.6 TB Data #1: Unaligned multi-frame micrographs of AcrB embedded in the liposome. [micrographs - multiframe])
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information
 Authors
Authors Citation
Citation
 Structure visualization
Structure visualization
 Downloads & links
Downloads & links emd_22050.png
emd_22050.png http://ftp.pdbj.org/pub/emdb/structures/EMD-22050
http://ftp.pdbj.org/pub/emdb/structures/EMD-22050










 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)




































 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN

