 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-13880 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the GPCR dimer Ste2 bound to an antagonist | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | GPCR / dimer / antagonist-bound / fungal / MEMBRANE PROTEIN | |||||||||
| Function / homology | mating-type factor pheromone receptor activity / GPCR fungal pheromone mating factor, STE2 / Pheromone alpha factor receptor, double transmembrane domain superfamily / Fungal pheromone mating factor STE2 GPCR / G protein-coupled receptor homodimeric complex / : / Pheromone alpha factor receptor Function and homology information Function and homology information | |||||||||
| Biological species |  | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 2.7 Å | |||||||||
 Authors Authors | Velazhahan V / Tate CG | |||||||||
| Funding support |  United Kingdom, 1 items United Kingdom, 1 items
| |||||||||
 Citation Citation | Journal: Nature / Year: 2021 Title: Structure of the class D GPCR Ste2 dimer coupled to two G proteins. Authors: Vaithish Velazhahan / Ning Ma / Gáspár Pándy-Szekeres / Albert J Kooistra / Yang Lee / David E Gloriam / Nagarajan Vaidehi / Christopher G Tate /    Abstract: G-protein-coupled receptors (GPCRs) are divided phylogenetically into six classes, denoted A to F. More than 370 structures of vertebrate GPCRs (belonging to classes A, B, C and F) have been ...G-protein-coupled receptors (GPCRs) are divided phylogenetically into six classes, denoted A to F. More than 370 structures of vertebrate GPCRs (belonging to classes A, B, C and F) have been determined, leading to a substantial understanding of their function. By contrast, there are no structures of class D GPCRs, which are found exclusively in fungi where they regulate survival and reproduction. Here we determine the structure of a class D GPCR, the Saccharomyces cerevisiae pheromone receptor Ste2, in an active state coupled to the heterotrimeric G protein Gpa1-Ste4-Ste18. Ste2 was purified as a homodimer coupled to two G proteins. The dimer interface of Ste2 is formed by the N terminus, the transmembrane helices H1, H2 and H7, and the first extracellular loop ECL1. We establish a class D1 generic residue numbering system (CD1) to enable comparisons with orthologues and with other GPCR classes. The structure of Ste2 bears similarities in overall topology to class A GPCRs, but the transmembrane helix H4 is shifted by more than 20 Å and the G-protein-binding site is a shallow groove rather than a cleft. The structure provides a template for the design of novel drugs to target fungal GPCRs, which could be used to treat numerous intractable fungal diseases. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_13880.map.gz | 5.9 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-13880-v30.xmlemd-13880.xml | 19.9 KB 19.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_13880_fsc.xml | 8.8 KB | Display | FSC data file |
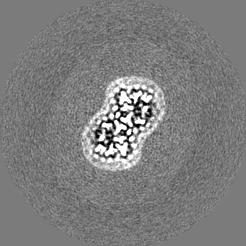
| Images | 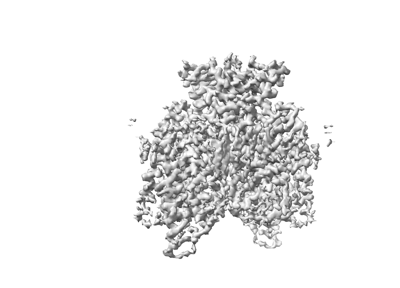 emd_13880.png emd_13880.png | 55.1 KB | ||
| Filedesc metadata | emd-13880.cif.gz | 6.4 KB | ||
| Others | emd_13880_half_map_1.map.gzemd_13880_half_map_2.map.gz | 42.6 MB 42.6 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-13880ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13880 http://ftp.pdbj.org/pub/emdb/structures/EMD-13880ftp://ftp.pdbj.org/pub/emdb/structures/EMD-13880 | HTTPS FTP |
-Related structure data
| Related structure data |  7qa8MC  7ad3C  7qb9C  7qbcC  7qbiC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10877 (Title: Structure of the GPCR dimer Ste2 bound to an antagonist Data size: 4.1 TB Data #1: unaligned multiframe micrographs of Ste2 in the antagonist-bound state [micrographs - multiframe]) |











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_13880.map.gz / Format: CCP4 / Size: 56.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.86 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
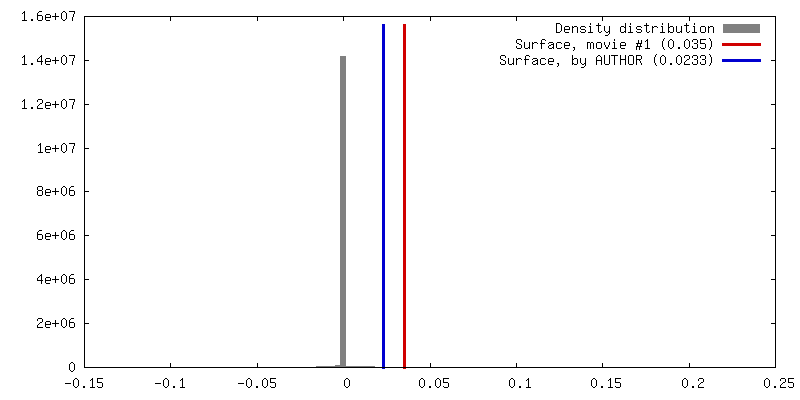
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: #2
| File | emd_13880_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
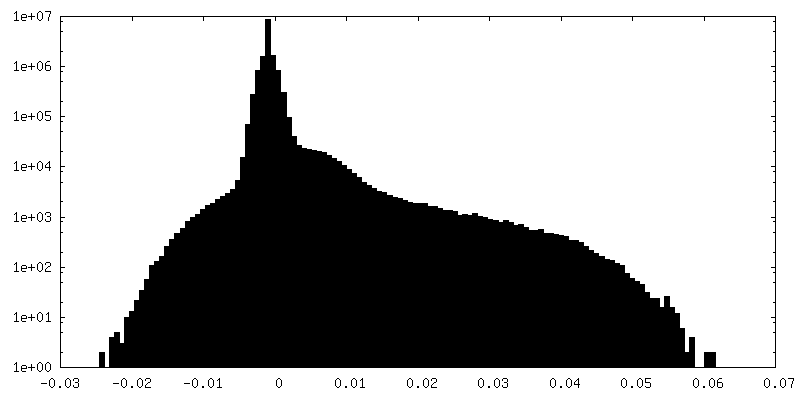
| Density Histograms |




-Half map: #1
| File | emd_13880_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
| Density Histograms |





- Sample components
Sample components
-Entire : GPCR dimer Ste2 bound to an antagonist
| Entire | Name: GPCR dimer Ste2 bound to an antagonist |
|---|---|
| Components |
|
-Supramolecule #1: GPCR dimer Ste2 bound to an antagonist
| Supramolecule | Name: GPCR dimer Ste2 bound to an antagonist / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#2 |
|---|---|
| Source (natural) | Organism: |
-Macromolecule #1: Pheromone alpha factor receptor
| Macromolecule | Name: Pheromone alpha factor receptor / type: protein_or_peptide / ID: 1 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 47.885402 KDa |
| Recombinant expression | Organism:  Trichoplusia ni (cabbage looper) Trichoplusia ni (cabbage looper) |
| Sequence | String: MSDAAPSLSN LFYDPTYNPG QSTINYTSIY GNGSTITFDE LQGLVNSTVT QAIMFGVRCG AAALTLIVMW MTSRSRKTPI FIINQVSLF LIILHSALYF KYLLSNYSSV TYALTGFPQF ISRGDVHVYG ATNIIQVLLV ASIETSLVFQ IKVIFTGDNF K RIGLMLTS ...String: MSDAAPSLSN LFYDPTYNPG QSTINYTSIY GNGSTITFDE LQGLVNSTVT QAIMFGVRCG AAALTLIVMW MTSRSRKTPI FIINQVSLF LIILHSALYF KYLLSNYSSV TYALTGFPQF ISRGDVHVYG ATNIIQVLLV ASIETSLVFQ IKVIFTGDNF K RIGLMLTS ISFTLGIATV TMYFVSAVKG MIVTYNDVSA TQDKYFNAST ILLASSINFM SFVLVVKLIL AIRSRRFLGL KQ FDSFHIL LIMSCQSLLV PSIIFILAYS LKPNQGTDVL TTVATLLAVL SLPLSSMWAT AANNASKTNT ITSDFTTSTD RFY PGTLSS FQTDSINNDA KSSLRSRLYD LYPRRKETTS DKHSERTFVS ETADDIEKNQ FYQLPTPTSS KNTRIGPFAD ASYK EGEVE PVDMYTPDTA ADEEARKFWT EDNNNL UniProtKB: Pheromone alpha factor receptor |
-Macromolecule #2: HIS-ALA-LEU-GLN-LEU-LYS-PRO-GLY-GLN-PRO-NLE-TYR
| Macromolecule | Name: HIS-ALA-LEU-GLN-LEU-LYS-PRO-GLY-GLN-PRO-NLE-TYR / type: protein_or_peptide / ID: 2 / Number of copies: 2 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: |
| Molecular weight | Theoretical: 1.366606 KDa |
| Sequence | String: HALQLKPGQP (NLE)Y |
-Macromolecule #3: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 3 / Number of copies: 4 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Macromolecule #4: CHOLESTEROL HEMISUCCINATE
| Macromolecule | Name: CHOLESTEROL HEMISUCCINATE / type: ligand / ID: 4 / Number of copies: 34 / Formula: Y01 |
|---|---|
| Molecular weight | Theoretical: 486.726 Da |
| Chemical component information |  ChemComp-Y01: |
-Macromolecule #5: water
| Macromolecule | Name: water / type: ligand / ID: 5 / Number of copies: 70 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 3.1 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | TFS KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 (6k x 4k) / Average electron dose: 57.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 50.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |

