 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7qa8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of the GPCR dimer Ste2 bound to an antagonist | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Components Components |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Keywords Keywords | MEMBRANE PROTEIN / GPCR / dimer / antagonist-bound / fungal | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Function / homology | mating-type factor pheromone receptor activity / GPCR fungal pheromone mating factor, STE2 / Pheromone alpha factor receptor, double transmembrane domain superfamily / Fungal pheromone mating factor STE2 GPCR / G protein-coupled receptor homodimeric complex / : / CHOLESTEROL HEMISUCCINATE / Pheromone alpha factor receptor Function and homology information Function and homology information | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Biological species |  | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Method | ELECTRON MICROSCOPY / single particle reconstruction / cryo EM / Resolution: 2.7 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Authors Authors | Velazhahan, V. / Tate, C.G. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Funding support |  United Kingdom, 1items United Kingdom, 1items
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Citation Citation | Journal: Nature / Year: 2021 Title: Structure of the class D GPCR Ste2 dimer coupled to two G proteins. Authors: Vaithish Velazhahan / Ning Ma / Gáspár Pándy-Szekeres / Albert J Kooistra / Yang Lee / David E Gloriam / Nagarajan Vaidehi / Christopher G Tate /    Abstract: G-protein-coupled receptors (GPCRs) are divided phylogenetically into six classes, denoted A to F. More than 370 structures of vertebrate GPCRs (belonging to classes A, B, C and F) have been ...G-protein-coupled receptors (GPCRs) are divided phylogenetically into six classes, denoted A to F. More than 370 structures of vertebrate GPCRs (belonging to classes A, B, C and F) have been determined, leading to a substantial understanding of their function. By contrast, there are no structures of class D GPCRs, which are found exclusively in fungi where they regulate survival and reproduction. Here we determine the structure of a class D GPCR, the Saccharomyces cerevisiae pheromone receptor Ste2, in an active state coupled to the heterotrimeric G protein Gpa1-Ste4-Ste18. Ste2 was purified as a homodimer coupled to two G proteins. The dimer interface of Ste2 is formed by the N terminus, the transmembrane helices H1, H2 and H7, and the first extracellular loop ECL1. We establish a class D1 generic residue numbering system (CD1) to enable comparisons with orthologues and with other GPCR classes. The structure of Ste2 bears similarities in overall topology to class A GPCRs, but the transmembrane helix H4 is shifted by more than 20 Å and the G-protein-binding site is a shallow groove rather than a cleft. The structure provides a template for the design of novel drugs to target fungal GPCRs, which could be used to treat numerous intractable fungal diseases. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | Molecule: MolmilJmol/JSmol |
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7qa8.cif.gz | 148.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7qa8.ent.gz | 118.6 KB | Display | PDB format |
| PDBx/mmJSON format | 7qa8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/qa/7qa8ftp://data.pdbj.org/pub/pdb/validation_reports/qa/7qa8 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  13880MC  7ad3C  7qb9C  7qbcC  7qbiC M: map data used to model this data C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10877 (Title: Structure of the GPCR dimer Ste2 bound to an antagonist Data size: 4.1 TB Data #1: unaligned multiframe micrographs of Ste2 in the antagonist-bound state [micrographs - multiframe]) |














-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
|
|---|---|
| 1 |
|
-Components
| #1: Protein | Mass: 47885.402 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Gene: STE2 / Production host:  Trichoplusia ni (cabbage looper) / References: UniProt: P0CI39 Trichoplusia ni (cabbage looper) / References: UniProt: P0CI39#2: Protein/peptide | Mass: 1366.606 Da / Num. of mol.: 2 / Source method: obtained synthetically / Source: (synth.) #3: Sugar | ChemComp-NAG /   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H15NO6 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C8H15NO6#4: Chemical | ChemComp-Y01 / 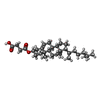  Mass: 486.726 Da / Num. of mol.: 34 / Source method: obtained synthetically / Formula: C31H50O4 Mass: 486.726 Da / Num. of mol.: 34 / Source method: obtained synthetically / Formula: C31H50O4#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 70 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: ELECTRON MICROSCOPY |
|---|---|
| EM experiment | Aggregation state: PARTICLE / 3D reconstruction method: single particle reconstruction |
- Sample preparation
Sample preparation
| Component | Name: GPCR dimer Ste2 bound to an antagonist / Type: COMPLEX / Entity ID: #1-#2 / Source: RECOMBINANT |
|---|---|
| Molecular weight | Experimental value: NO |
| Source (natural) | Organism: |
| Source (recombinant) | Organism: Trichoplusia ni (cabbage looper) |
| Buffer solution | pH: 7.5 |
| Specimen | Conc.: 3.1 mg/ml / Embedding applied: NO / Shadowing applied: NO / Staining applied: NO / Vitrification applied: YES |
| Vitrification | Instrument: FEI VITROBOT MARK IV / Cryogen name: ETHANE / Humidity: 100 % / Chamber temperature: 277 K |
- Electron microscopy imaging
Electron microscopy imaging
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
|---|---|
| Microscopy | Model: TFS KRIOS |
| Electron gun | Electron source:  FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM FIELD EMISSION GUN / Accelerating voltage: 300 kV / Illumination mode: FLOOD BEAM |
| Electron lens | Mode: BRIGHT FIELD / Cs: 2.7 mm / C2 aperture diameter: 50 µm |
| Image recording | Electron dose: 57 e/Å2 / Film or detector model: GATAN K3 (6k x 4k) |
- Processing
Processing
| Software | Name: PHENIX / Version: 1.19.2_4158: / Classification: refinement | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EM software |
| ||||||||||||||||||||||||
| CTF correction | Type: NONE | ||||||||||||||||||||||||
| Symmetry | Point symmetry: C2 (2 fold cyclic) | ||||||||||||||||||||||||
| 3D reconstruction | Resolution: 2.7 Å / Resolution method: FSC 0.143 CUT-OFF / Num. of particles: 136877 / Symmetry type: POINT | ||||||||||||||||||||||||
| Refine LS restraints |
|
