 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-10649: Cryo-EM structure of the prefusion state of canine distemper viru... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-10649 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
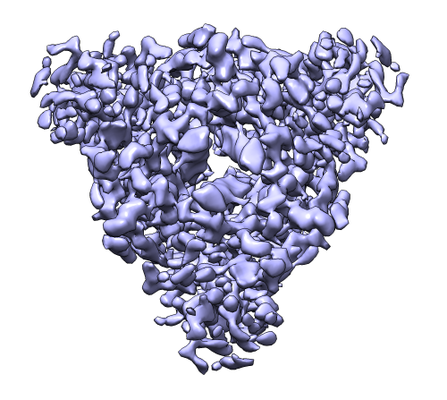
| Title | Cryo-EM structure of the prefusion state of canine distemper virus fusion protein ectodomain | |||||||||
 Map data Map data | ||||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | fusion protein / prefusion state / VIRAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationfusion of virus membrane with host plasma membrane / viral envelope / symbiont entry into host cell / host cell plasma membrane / virion membrane / plasma membrane Similarity search - Function | |||||||||
| Biological species |  Canine morbillivirus Canine morbillivirus | |||||||||
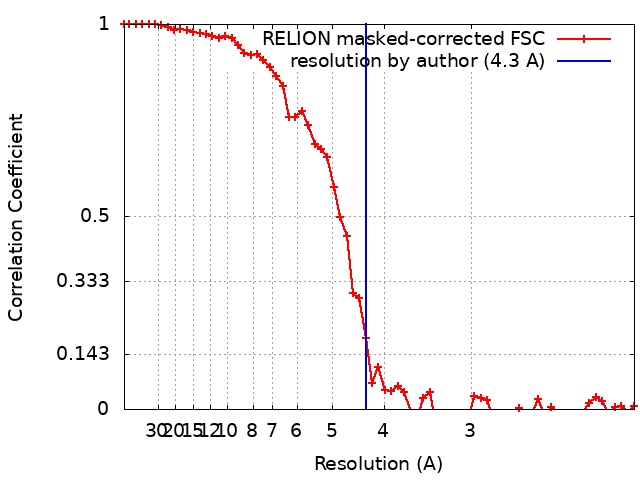
| Method | single particle reconstruction / cryo EM / Resolution: 4.3 Å | |||||||||
 Authors Authors | Kalbermatter D / Fotiadis D | |||||||||
| Funding support |  Switzerland, 1 items Switzerland, 1 items
| |||||||||
 Citation Citation | Journal: J Struct Biol X / Year: 2020 Title: Cryo-EM structure of the prefusion state of canine distemper virus fusion protein ectodomain. Authors: David Kalbermatter / Neeta Shrestha / Flavio M Gall / Marianne Wyss / Rainer Riedl / Philippe Plattet / Dimitrios Fotiadis / Abstract: Measles virus (MeV) and canine distemper virus (CDV), two members of the genus, are still causing important global diseases of humans and animals, respectively. To enter target cells, ...Measles virus (MeV) and canine distemper virus (CDV), two members of the genus, are still causing important global diseases of humans and animals, respectively. To enter target cells, morbilliviruses rely on an envelope-anchored machinery, which is composed of two interacting glycoproteins: a tetrameric receptor binding (H) protein and a trimeric fusion (F) protein. To execute membrane fusion, the F protein initially adopts a metastable, prefusion state that refolds into a highly stable postfusion conformation as the result of a finely coordinated activation process mediated by the H protein. Here, we employed cryo-electron microscopy (cryo-EM) and single particle reconstruction to elucidate the structure of the prefusion state of the CDV F protein ectodomain (solF) at 4.3 Å resolution. Stabilization of the prefusion solF trimer was achieved by fusing the GCNt trimerization sequence at the C-terminal protein region, and expressing and purifying the recombinant protein in the presence of a morbilliviral fusion inhibitor class compound. The three-dimensional cryo-EM map of prefusion CDV solF in complex with the inhibitor clearly shows density for the ligand at the protein binding site suggesting common mechanisms of membrane fusion activation and inhibition employed by different morbillivirus members. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_10649.map.gz | 2.3 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-10649-v30.xmlemd-10649.xml | 19.3 KB 19.3 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_10649_fsc.xml | 5.8 KB | Display | FSC data file |
| Images |  emd_10649.png emd_10649.png | 184.7 KB | ||
| Filedesc metadata | emd-10649.cif.gz | 6.1 KB | ||
| Others | emd_10649_half_map_1.map.gzemd_10649_half_map_2.map.gz | 11.9 MB 11.9 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-10649ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10649 http://ftp.pdbj.org/pub/emdb/structures/EMD-10649ftp://ftp.pdbj.org/pub/emdb/structures/EMD-10649 | HTTPS FTP |
-Related structure data
| Related structure data |  6xyeMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data | |
| EM raw data | EMPIAR-10366 (Title: Cryo-EM structure of the prefusion state of canine distemper virus fusion protein ectodomain Data size: 474.1 Data #1: Unaligned multi-frame micrographs of the prefusion state of canine distemper virus fusion protein ectodomain. [micrographs - multiframe]) |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_10649.map.gz / Format: CCP4 / Size: 15.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.021 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: half map 1
| File | emd_10649_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |

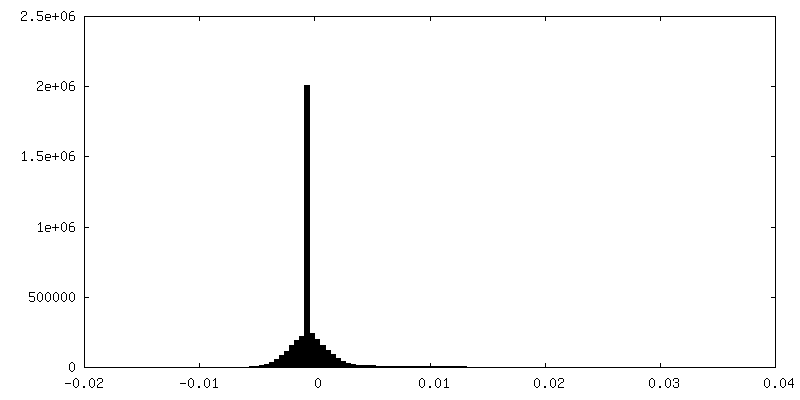
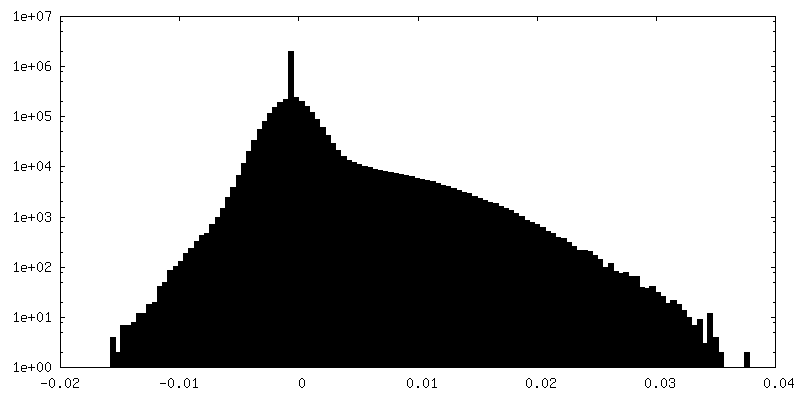
-Half map: half map 2
| File | emd_10649_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | half map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |



- Sample components
Sample components
-Entire : Canine distemper virus fusion protein
| Entire | Name: Canine distemper virus fusion protein |
|---|---|
| Components |
|
-Supramolecule #1: Canine distemper virus fusion protein
| Supramolecule | Name: Canine distemper virus fusion protein / type: complex / ID: 1 / Parent: 0 / Macromolecule list: all |
|---|---|
| Source (natural) | Organism: Canine morbillivirus |
-Macromolecule #1: Fusion glycoprotein F2
| Macromolecule | Name: Fusion glycoprotein F2 / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Canine morbillivirus |
| Molecular weight | Theoretical: 10.128781 KDa |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Sequence | String: QIHWNNLSTI GIIGTDSVHY KIMTRPSHQY LVIKLMPNVS LIDNCTKAEL GEYEKLLNSV LEPINQALTL MTKNVKPLQS VGSGRRQRR UniProtKB: Fusion glycoprotein F0 |
-Macromolecule #2: Fusion glycoprotein F1
| Macromolecule | Name: Fusion glycoprotein F1 / type: protein_or_peptide / ID: 2 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Canine morbillivirus |
| Molecular weight | Theoretical: 47.511637 KDa |
| Recombinant expression | Organism: Homo sapiens (human) |
| Sequence | String: FAGVVLAGAA LGVATAAQIT AGIALHQSNL NAQAIQSLRT SLEQSNKAIE EIREATQETV IAVQGVQDYV NNELVPAMQH MSCELVGQR LGLKLLRYYT ELLSIFGPSL RDPISAEISI QALSYALGGE IHKILEKLGY SGNDMIAILE SRGIKTKITH V DLPGKLII ...String: FAGVVLAGAA LGVATAAQIT AGIALHQSNL NAQAIQSLRT SLEQSNKAIE EIREATQETV IAVQGVQDYV NNELVPAMQH MSCELVGQR LGLKLLRYYT ELLSIFGPSL RDPISAEISI QALSYALGGE IHKILEKLGY SGNDMIAILE SRGIKTKITH V DLPGKLII LSISYPTLSE VKGVIVHRLE AVSYNIGSQE WYTTVPRYVA TNGYLISNFD ESPCVFVSES AICSQNSLYP MS PLLQQCI RGDTSSCART LVSGTMGNKF ILSKGNIVAN CASILCKCYS TGTIINQSPD KLLTFIASDT CPLVEIDGVT IQV GGRQYP DMVYESKVAL GPAISLERLD VGTNLGNALK KLDDAKVLID SSNQILETVR RSSFNFGSLL SVPILICTAL ALLL LIYCC KRRYQQTLKQ NAKVDPTFKP DLTGTSKSYV RSL UniProtKB: Fusion glycoprotein F0 |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.23 mg/mL | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| |||||||||||||||
| Grid | Model: Quantifoil R2/1 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 120 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.025 kPa | |||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277.15 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Number grids imaged: 1 / Number real images: 1604 / Average electron dose: 73.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model |
| ||||||
|---|---|---|---|---|---|---|---|
| Refinement | Space: REAL / Protocol: RIGID BODY FIT | ||||||
| Output model | PDB-6xye: |

