 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-8659: VCP like ATPase from T. acidophilum (VAT) - Substrate bound confo... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8659 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
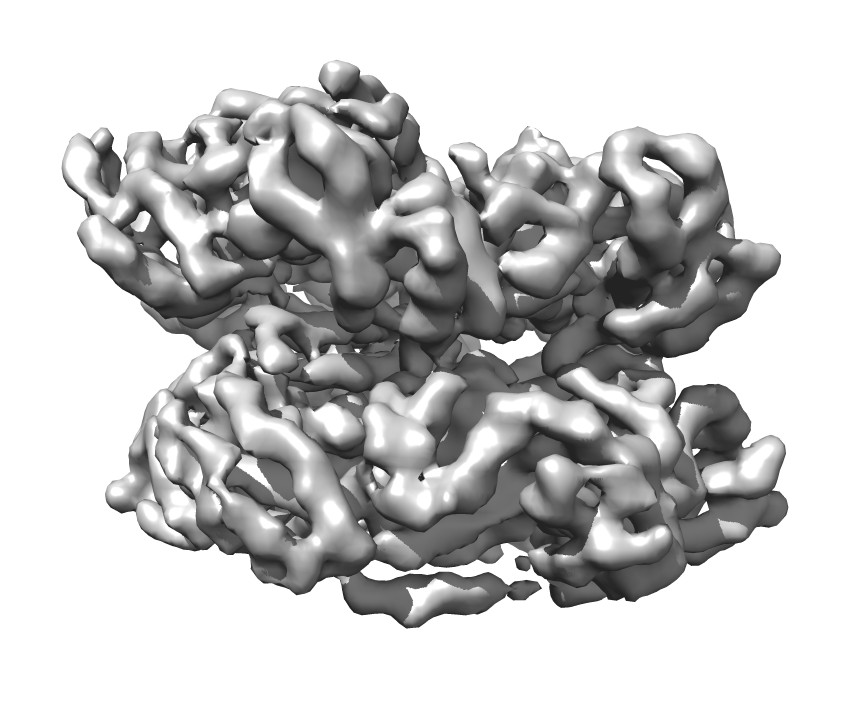
| Title | VCP like ATPase from T. acidophilum (VAT) - Substrate bound conformation | |||||||||
 Map data Map data | VCP like ATPase from T. acidophilum - Substrate engaged state | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | AAA+ / ATPase / Complex / unfoldase / HYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology information | |||||||||
| Biological species |   Thermoplasma acidophilum (acidophilic) / Thermoplasma acidophilum (strain ATCC 25905 / DSM 1728 / JCM 9062 / NBRC 15155 / AMRC-C165) (acidophilic) Thermoplasma acidophilum (acidophilic) / Thermoplasma acidophilum (strain ATCC 25905 / DSM 1728 / JCM 9062 / NBRC 15155 / AMRC-C165) (acidophilic) | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.8 Å | |||||||||
 Authors Authors | Ripstein ZA / Huang R | |||||||||
| Funding support |  Canada, 2 items Canada, 2 items
| |||||||||
 Citation Citation | Journal: Elife / Year: 2017 Title: Structure of a AAA+ unfoldase in the process of unfolding substrate. Authors: Zev A Ripstein / Rui Huang / Rafal Augustyniak / Lewis E Kay / John L Rubinstein / Abstract: AAA+ unfoldases are thought to unfold substrate through the central pore of their hexameric structures, but how this process occurs is not known. VAT, the homologue of eukaryotic CDC48/p97, works in ...AAA+ unfoldases are thought to unfold substrate through the central pore of their hexameric structures, but how this process occurs is not known. VAT, the homologue of eukaryotic CDC48/p97, works in conjunction with the proteasome to degrade misfolded or damaged proteins. We show that in the presence of ATP, VAT with its regulatory N-terminal domains removed unfolds other VAT complexes as substrate. We captured images of this transient process by electron cryomicroscopy (cryo-EM) to reveal the structure of the substrate-bound intermediate. Substrate binding breaks the six-fold symmetry of the complex, allowing five of the six VAT subunits to constrict into a tight helix that grips an ~80 Å stretch of unfolded protein. The structure suggests a processive hand-over-hand unfolding mechanism, where each VAT subunit releases the substrate in turn before re-engaging further along the target protein, thereby unfolding it. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8659.map.gz | 59.6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8659-v30.xmlemd-8659.xml | 14.5 KB 14.5 KB | Display Display | EMDB header |
| Images |  emd_8659.png emd_8659.png | 279.4 KB | ||
| Filedesc metadata | emd-8659.cif.gz | 6.3 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8659ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8659 http://ftp.pdbj.org/pub/emdb/structures/EMD-8659ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8659 | HTTPS FTP |
-Validation report
| Summary document | emd_8659_validation.pdf.gz | 552.1 KB | Display | EMDB validaton report |
|---|---|---|---|---|
| Full document | emd_8659_full_validation.pdf.gz | 551.6 KB | Display | |
| Data in XML | emd_8659_validation.xml.gz | 6.1 KB | Display | |
| Data in CIF | emd_8659_validation.cif.gz | 7.1 KB | Display | |
| Arichive directory | https://ftp.pdbj.org/pub/emdb/validation_reports/EMD-8659ftp://ftp.pdbj.org/pub/emdb/validation_reports/EMD-8659 | HTTPS FTP |
-Related structure data
| Related structure data |  5vcaMC  8658C  5vc7C C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |






-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |




-Map
| File | Download / File: emd_8659.map.gz / Format: CCP4 / Size: 64 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | VCP like ATPase from T. acidophilum - Substrate engaged state | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
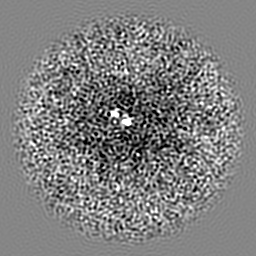
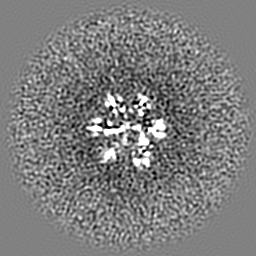
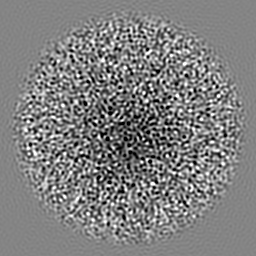
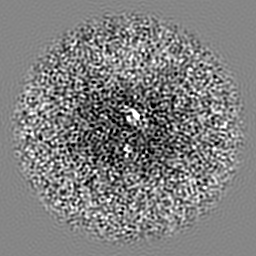
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.45 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
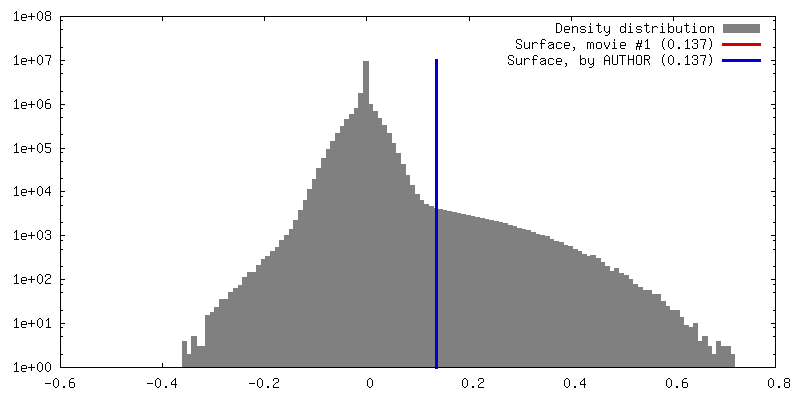
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)












-Supplemental data
- Sample components
Sample components
-Entire : Structural basis for substrate unfolding by the VCP like ATPase f...
| Entire | Name: Structural basis for substrate unfolding by the VCP like ATPase from T. Acidophilum |
|---|---|
| Components |
|
-Supramolecule #1: Structural basis for substrate unfolding by the VCP like ATPase f...
| Supramolecule | Name: Structural basis for substrate unfolding by the VCP like ATPase from T. Acidophilum type: complex / ID: 1 / Parent: 0 / Macromolecule list: all Details: Stacked-ring state, ATPgS loaded N-terminal domain removed |
|---|---|
| Source (natural) | Organism: Thermoplasma acidophilum (acidophilic) |
| Molecular weight | Theoretical: 380 KDa |
-Macromolecule #1: VCP-like ATPase
| Macromolecule | Name: VCP-like ATPase / type: protein_or_peptide / ID: 1 / Number of copies: 6 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Thermoplasma acidophilum (strain ATCC 25905 / DSM 1728 / JCM 9062 / NBRC 15155 / AMRC-C165) (acidophilic) Strain: ATCC 25905 / DSM 1728 / JCM 9062 / NBRC 15155 / AMRC-C165 |
| Molecular weight | Theoretical: 62.996246 KDa |
| Recombinant expression | Organism:  |
| Sequence | String: MEVSRISYED IGGLSEQLGK IREMIELPLK HPELFERLGI TPPKGVILYG PPGTGKTLIA RAVANESGAN FLSINGPEIM SKYYGQSEQ KLREIFSKAE ETAPSIIFID EIDSIAPKRE EVQGEVERRV VAQLLTLMDG MKERGHVIVI GATNRIDAID P ALRRPGRF ...String: MEVSRISYED IGGLSEQLGK IREMIELPLK HPELFERLGI TPPKGVILYG PPGTGKTLIA RAVANESGAN FLSINGPEIM SKYYGQSEQ KLREIFSKAE ETAPSIIFID EIDSIAPKRE EVQGEVERRV VAQLLTLMDG MKERGHVIVI GATNRIDAID P ALRRPGRF DREIEIGVPD RNGRKEILMI HTRNMPLGMS EEEKNKFLEE MADYTYGFVG ADLAALVRES AMNALRRYLP EI DLDKPIP TEILEKMVVT EDDFKNALKS IEPSSLREVM VEVPNVHWDD IGGLEDVKRE IKETVELPLL KPDVFKRLGI RPS KGFLLY GPPGVGKTLL AKAVATESNA NFISIKGPEV LSKWVGESEK AIREIFKKAK QVAPAIVFLD EIDSIAPRRG TTSD SGVTE RIVNQLLTSL DGIEVMNGVV VIGATNRPDI MDPALLRAGR FDKLIYIPPP DKEARLSILK VHTKNMPLAP DVDLN DIAQ RTEGYVGADL ENLCREAGMN AYRENPDATS VSQKNFLDAL KTIRPSVDEE VIKFYRTLSE TMSKSVSERR KQLQDQ GLY L UniProtKB: VCP-like ATPase |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 20 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Electron Microscopy Sciences M400 / Material: COPPER/RHODIUM / Mesh: 400 / Support film - topology: HOLEY ARRAY / Support film - Film thickness: 30 |
| Vitrification | Cryogen name: ETHANE-PROPANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK III / Details: Blot for 4 seconds before plunging. |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI 20 |
|---|---|
| Image recording | Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3838 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Frames/image: 1-30 / Number grids imaged: 3 / Number real images: 246 / Average exposure time: 15.0 sec. / Average electron dose: 35.0 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 30.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.0 mm / Nominal defocus max: 2.9 µm / Nominal defocus min: 1.7 µm / Nominal magnification: 25000 |
| Sample stage | Specimen holder model: GATAN 626 SINGLE TILT LIQUID NITROGEN CRYO TRANSFER HOLDER Cooling holder cryogen: NITROGEN |
