 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7sjb: Crystal structure of dehaloperoxidase B in complex with alpha-ter... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7sjb | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of dehaloperoxidase B in complex with alpha-terpinene | ||||||
 Components Components | Dehaloperoxidase B | ||||||
 Keywords Keywords | OXIDOREDUCTASE / heme peroxidase / peroxygenase / heme cofactor / oxygen binding | ||||||
| Function / homology |  Function and homology information Function and homology informationoxygen carrier activity / peroxidase activity / oxygen binding / heme binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   Amphitrite ornata (invertebrata) Amphitrite ornata (invertebrata) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.718 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.718 Å | ||||||
 Authors Authors | Ghiladi, R.A. / de Serrano, V.S. / Malewschik, T. / Yun, D. | ||||||
| Funding support |  United States, 1items United States, 1items
| ||||||
 Citation Citation | Journal: To Be Published Title: The Multifunctional Globin Dehaloperoxidase as a Biocatalyst in the Oxidation of Monoterpenes Authors: Malewschick, T. / Yun, D. / de Serrano, V. / Ghiladi, R.A. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7sjb.cif.gz | 132.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7sjb.ent.gz | 100.1 KB | Display | PDB format |
| PDBx/mmJSON format | 7sjb.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7sjb_validation.pdf.gz | 1.5 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7sjb_full_validation.pdf.gz | 1.5 MB | Display | |
| Data in XML | 7sjb_validation.xml.gz | 17.1 KB | Display | |
| Data in CIF | 7sjb_validation.cif.gz | 22.6 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sj/7sjbftp://data.pdbj.org/pub/pdb/validation_reports/sj/7sjb | HTTPS FTP |
-Related structure data
| Related structure data |  7sjcC  7sjdC  7sjeC  7sjfC  7sjgC  7sjhC  7sjiC  7t9cC  7t9dC  7t9eC  3ixfS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |
-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 2 molecules AB
| #1: Protein | Mass: 15414.462 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Amphitrite ornata (invertebrata) / Plasmid: pET28a / Production host:  |
|---|
-Non-polymers , 5 types, 111 molecules 
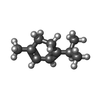



| #2: Chemical |  Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4 / Feature type: SUBJECT OF INVESTIGATION#3: Chemical |  Mass: 136.234 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16 / Feature type: SUBJECT OF INVESTIGATION Mass: 136.234 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H16 / Feature type: SUBJECT OF INVESTIGATION#4: Chemical |  Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: SO4#5: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 104 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Sequence details | Ligand 9MI occupies density that is close to the position of H55 in its alternate conformation. The ...Ligand 9MI occupies density that is close to the position of H55 in its alternate conformation. The occupancies of the alternate conformations reflect the fact. In the fraction where the ligand is bound, reflected by the occupancy of the ligand, H55 is in the conformation facing outside of the distal side of the heme pocket, with the same occupancy as the bound ligand. When the ligand is not bound, H55 can occupy the inside conformation, also reflected by its occupancy. |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.24 Å3/Da / Density % sol: 45.15 % |
|---|---|
| Crystal grow | Temperature: 277.15 K / Method: vapor diffusion, hanging drop / pH: 6.5 / Details: MPEG 2000, ammonium sulfate, Na cacodylate |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS / Beamline: 22-BM / Wavelength: 1 Å | ||||||||||||||||||
| Detector | Type: RAYONIX MX300-HS / Detector: CCD / Date: Sep 22, 2021 | ||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||
| Reflection twin |
| ||||||||||||||||||
| Reflection | Resolution: 1.718→33.77 Å / Num. obs: 30039 / % possible obs: 99.4 % / Redundancy: 5 % / CC1/2: 0.996 / Rmerge(I) obs: 0.056 / Net I/σ(I): 31.2 | ||||||||||||||||||
| Reflection shell | Resolution: 1.72→1.762 Å / Num. unique obs: 1953 / CC1/2: 0.803 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3IXF Resolution: 1.718→33.766 Å / Cor.coef. Fo:Fc: 0.972 / Cor.coef. Fo:Fc free: 0.956 / SU B: 3.124 / SU ML: 0.057 / Cross valid method: THROUGHOUT / ESU R: 0.024 / ESU R Free: 0.023 Details: Hydrogens have been added in their riding positions
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK BULK SOLVENT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.651 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.718→33.766 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
