 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7efl: Crystal structure of the gastric proton pump K791S in (BYK)E2BeF state -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7efl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the gastric proton pump K791S in (BYK)E2BeF state | ||||||
 Components Components |
| ||||||
 Keywords Keywords | MEMBRANE PROTEIN / Cation pump / P-type ATPase / gastric / proton pump | ||||||
| Function / homology |  Function and homology information Function and homology informationpotassium:proton exchanging ATPase complex / P-type potassium:proton transporter activity / Ion transport by P-type ATPases / sodium:potassium-exchanging ATPase complex / sodium ion export across plasma membrane / intracellular sodium ion homeostasis / potassium ion import across plasma membrane / ATPase activator activity / intracellular potassium ion homeostasis / potassium ion transmembrane transport ...potassium:proton exchanging ATPase complex / P-type potassium:proton transporter activity / Ion transport by P-type ATPases / sodium:potassium-exchanging ATPase complex / sodium ion export across plasma membrane / intracellular sodium ion homeostasis / potassium ion import across plasma membrane / ATPase activator activity / intracellular potassium ion homeostasis / potassium ion transmembrane transport / proton transmembrane transport / cell adhesion / apical plasma membrane / magnesium ion binding / ATP hydrolysis activity / ATP binding Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.4 Å | ||||||
 Authors Authors | Abe, K. / Yamamoto, K. / Irie, K. | ||||||
| Funding support |  Japan, 1items Japan, 1items
| ||||||
 Citation Citation | Journal: Nat Commun / Year: 2021 Title: Gastric proton pump with two occluded K engineered with sodium pump-mimetic mutations. Authors: Kazuhiro Abe / Kenta Yamamoto / Katsumasa Irie / Tomohiro Nishizawa / Atsunori Oshima / Abstract: The gastric H,K-ATPase mediates electroneutral exchange of 1H/1K per ATP hydrolysed across the membrane. Previous structural analysis of the K-occluded E2-P transition state of H,K-ATPase showed a ...The gastric H,K-ATPase mediates electroneutral exchange of 1H/1K per ATP hydrolysed across the membrane. Previous structural analysis of the K-occluded E2-P transition state of H,K-ATPase showed a single bound K at cation-binding site II, in marked contrast to the two K ions occluded at sites I and II of the closely-related Na,K-ATPase which mediates electrogenic 3Na/2K translocation across the membrane. The molecular basis of the different K stoichiometry between these K-counter-transporting pumps is elusive. We show a series of crystal structures and a cryo-EM structure of H,K-ATPase mutants with changes in the vicinity of site I, based on the structure of the sodium pump. Our step-wise and tailored construction of the mutants finally gave a two-K bound H,K-ATPase, achieved by five mutations, including amino acids directly coordinating K (Lys791Ser, Glu820Asp), indirectly contributing to cation-binding site formation (Tyr340Asn, Glu936Val), and allosterically stabilizing K-occluded conformation (Tyr799Trp). This quintuple mutant in the K-occluded E2-P state unambiguously shows two separate densities at the cation-binding site in its 2.6 Å resolution cryo-EM structure. These results offer new insights into how two closely-related cation pumps specify the number of K accommodated at their cation-binding site. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7efl.cif.gz | 569.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7efl.ent.gz | 416 KB | Display | PDB format |
| PDBx/mmJSON format | 7efl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ef/7eflftp://data.pdbj.org/pub/pdb/validation_reports/ef/7efl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  7efmC  7efnC  7et1C  5ylvS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
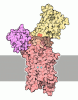
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
|
-Components
-Protein , 2 types, 2 molecules AB
| #1: Protein | Mass: 109220.570 Da / Num. of mol.: 1 / Mutation: K791S Source method: isolated from a genetically manipulated source Source: (gene. exp.)  Homo sapiens (human) / References: UniProt: A0A5G2QYH2 Homo sapiens (human) / References: UniProt: A0A5G2QYH2 |
|---|---|
| #2: Protein | Mass: 28976.170 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / References: UniProt: P18434 |
-Sugars , 1 types, 2 molecules 
| #6: Sugar |  Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H15NO6 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H15NO6 |
|---|
-Non-polymers , 4 types, 9 molecules 

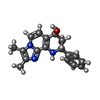

| #3: Chemical | ChemComp-MG /  Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg | ||||
|---|---|---|---|---|---|
| #4: Chemical | ChemComp-RB /  Mass: 85.468 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Rb Mass: 85.468 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Rb#5: Chemical | ChemComp-J3C / ( |  Mass: 309.362 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H19N3O2 / Feature type: SUBJECT OF INVESTIGATION Mass: 309.362 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C18H19N3O2 / Feature type: SUBJECT OF INVESTIGATION#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 3 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.26 Å3/Da / Density % sol: 71.11 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 10% glycerol, 20% PEG 2000 MME, 0.2M KCl, 3% methylpentanediol, 5mM beta-mercaptoethanol |
-Data collection
| Diffraction | Mean temperature: 80 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SPring-8 / Beamline: BL41XU / Wavelength: 1 Å |
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Nov 28, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.4→48.29 Å / Num. obs: 33564 / % possible obs: 99.8 % / Redundancy: 20 % / Biso Wilson estimate: 94.55 Å2 / CC1/2: 0.99 / Rmerge(I) obs: 0.165 / Rpim(I) all: 0.039 / Net I/σ(I): 12.4 |
| Reflection shell | Resolution: 3.4→3.5 Å / Redundancy: 21.1 % / Rmerge(I) obs: 2.48 / Num. unique obs: 3274 / CC1/2: 0.79 / Rpim(I) all: 0.548 / % possible all: 99.8 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5ylv Resolution: 3.4→48.29 Å / SU ML: 0.4588 / Cross valid method: FREE R-VALUE / Phase error: 32.3601 Stereochemistry target values: GeoStd + Monomer Library + CDL v1.2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 103.43 Å2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.4→48.29 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group | Refine-ID: X-RAY DIFFRACTION
|
