 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-7buc: Crystal structure of EHMT2 SET domain in complex with compound 13 -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 7buc | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of EHMT2 SET domain in complex with compound 13 | ||||||
 Components Components | Histone-lysine N-methyltransferase EHMT2 | ||||||
 Keywords Keywords | TRANSFERASE/TRANSFERASE INHIBITOR / PROTEIN-SMALL MOLECULE INHIBITOR COMPLEX / TRANSFERASE-TRANSFERASE INHIBITOR COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of protein modification process / histone H3K56 methyltransferase activity / phenotypic switching / neuron fate specification / [histone H3]-lysine9 N-methyltransferase / peptidyl-lysine dimethylation / histone H3K27 methyltransferase activity / histone H3K9 methyltransferase activity / histone H3K9me2 methyltransferase activity / synaptonemal complex assembly ...regulation of protein modification process / histone H3K56 methyltransferase activity / phenotypic switching / neuron fate specification / [histone H3]-lysine9 N-methyltransferase / peptidyl-lysine dimethylation / histone H3K27 methyltransferase activity / histone H3K9 methyltransferase activity / histone H3K9me2 methyltransferase activity / synaptonemal complex assembly / negative regulation of autophagosome assembly / oocyte development / protein-lysine N-methyltransferase activity / C2H2 zinc finger domain binding / fertilization / DNA methylation-dependent constitutive heterochromatin formation / cellular response to cocaine / organ growth / negative regulation of gene expression via chromosomal CpG island methylation / regulation of DNA replication / Transcriptional Regulation by E2F6 / RNA Polymerase I Transcription Initiation / behavioral response to cocaine / Transcriptional Regulation by VENTX / spermatid development / long-term memory / response to fungicide / cellular response to starvation / Regulation of endogenous retroelements by the Human Silencing Hub (HUSH) complex / transcription corepressor binding / Transferases; Transferring one-carbon groups; Methyltransferases / ERCC6 (CSB) and EHMT2 (G9a) positively regulate rRNA expression / promoter-specific chromatin binding / RNA polymerase II transcription regulatory region sequence-specific DNA binding / Regulation of TP53 Activity through Methylation / PKMTs methylate histone lysines / p53 binding / cellular response to xenobiotic stimulus / Senescence-Associated Secretory Phenotype (SASP) / response to ethanol / nuclear speck / chromatin / negative regulation of transcription by RNA polymerase II / enzyme binding / zinc ion binding / nucleoplasm / nucleus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Suzuki, M. / Mizuno, M. / Katayama, K. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2020 Title: Discovery of novel histone lysine methyltransferase G9a/GLP (EHMT2/1) inhibitors: Design, synthesis, and structure-activity relationships of 2,4-diamino-6-methylpyrimidines. Authors: Katayama, K. / Ishii, K. / Tsuda, E. / Yotsumoto, K. / Hiramoto, K. / Suzuki, M. / Yasumatsu, I. / Igarashi, W. / Torihata, M. / Ishiyama, T. / Katagiri, T. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 7buc.cif.gz | 126.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb7buc.ent.gz | 94.5 KB | Display | PDB format |
| PDBx/mmJSON format | 7buc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 7buc_validation.pdf.gz | 1.5 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 7buc_full_validation.pdf.gz | 1.5 MB | Display | |
| Data in XML | 7buc_validation.xml.gz | 22 KB | Display | |
| Data in CIF | 7buc_validation.cif.gz | 29.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bu/7bucftp://data.pdbj.org/pub/pdb/validation_reports/bu/7buc | HTTPS FTP |
-Related structure data
| Related structure data |  7btvC  5vseS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj














- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 32604.924 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: EHMT2, BAT8, C6orf30, G9A, KMT1C, NG36 / Plasmid: pET15b / Production host:  References: UniProt: Q96KQ7, Transferases; Transferring one-carbon groups; Methyltransferases #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Zn#3: Chemical | 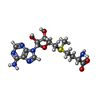  Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S Mass: 398.437 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C15H22N6O5S#4: Chemical | 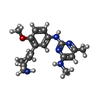  Mass: 339.435 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H25N5O / Feature type: SUBJECT OF INVESTIGATION Mass: 339.435 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H25N5O / Feature type: SUBJECT OF INVESTIGATION#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2OHas ligand of interest | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.38 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop Details: 0.2 M magnesium acetate, 20% polyethyleneglycol 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SLS  / Beamline: X10SA / Wavelength: 1 Å / Beamline: X10SA / Wavelength: 1 Å | ||||||||||||||||||||||||||||||
| Detector | Type: DECTRIS EIGER X 16M / Detector: PIXEL / Date: Sep 21, 2018 | ||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||||||||||||
| Reflection | Resolution: 2.6→78.43 Å / Num. obs: 19580 / % possible obs: 99.3 % / Redundancy: 3.1 % / CC1/2: 0.992 / Rmerge(I) obs: 0.116 / Rpim(I) all: 0.078 / Rrim(I) all: 0.141 / Net I/σ(I): 6.8 / Num. measured all: 60135 / Scaling rejects: 21 | ||||||||||||||||||||||||||||||
| Reflection shell | Diffraction-ID: 1
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 5VSE Resolution: 2.6→25 Å / Cor.coef. Fo:Fc: 0.947 / Cor.coef. Fo:Fc free: 0.903 / SU B: 17.319 / SU ML: 0.327 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 1.042 / ESU R Free: 0.335 / Details: U VALUES : REFINED INDIVIDUALLY
| |||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å | |||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 111.49 Å2 / Biso mean: 48.87 Å2 / Biso min: 24.32 Å2
| |||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2.6→25 Å
| |||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.69 Å / Rfactor Rfree error: 0 / Total num. of bins used: 15
|
