Entry Database : PDB / ID : 6t2wTitle Crystal structure of the CSF1R kinase domain with a dihydropurinone inhibitor (compound 4) Macrophage colony-stimulating factor 1 receptor Keywords / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / / / Resolution : 1.7 Å Authors Schimpl, M. / Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Howard, M.R. / Williamson, B. ...Schimpl, M. / Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Howard, M.R. / Williamson, B. / Davies, B.R. / Cadogan, E.B. / Ramos-Montoya, A. / Dean, E. Journal : J.Med.Chem. / Year : 2020Title: The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-pyran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein ... Title : The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-pyran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor.Authors: Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Schimpl, M. / Howard, M.R. / Williamson, B. / Vazquez-Chantada, M. / Barratt, D.G. ... Authors : Goldberg, F.W. / Finlay, M.R.V. / Ting, A.K.T. / Beattie, D. / Lamont, G.M. / Fallan, C. / Wrigley, G.L. / Schimpl, M. / Howard, M.R. / Williamson, B. / Vazquez-Chantada, M. / Barratt, D.G. / Davies, B.R. / Cadogan, E.B. / Ramos-Montoya, A. / Dean, E. History Deposition Oct 9, 2019 Deposition site / Processing site Revision 1.0 Jan 1, 2020 Provider / Type Revision 1.1 Jan 22, 2020 Group / Category / citation_authorItem / _citation.year / _citation_author.nameRevision 1.2 Apr 22, 2020 Group / Category / citation_authorItem _citation.journal_volume / _citation.page_first ... _citation.journal_volume / _citation.page_first / _citation.page_last / _citation_author.identifier_ORCID Revision 1.3 May 1, 2024 Group / Database references / Refinement descriptionCategory chem_comp_atom / chem_comp_bond ... chem_comp_atom / chem_comp_bond / database_2 / pdbx_initial_refinement_model Item / _database_2.pdbx_database_accession
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj














 Assembly
Assembly

 Spodoptera frugiperda (fall armyworm)
Spodoptera frugiperda (fall armyworm)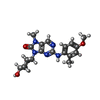
 Mass: 383.444 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H25N5O3 / Feature type: SUBJECT OF INVESTIGATION
Mass: 383.444 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H25N5O3 / Feature type: SUBJECT OF INVESTIGATION
 Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4
Mass: 96.063 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: SO4 Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: I03 / Wavelength: 0.97626 Å
/ Beamline: I03 / Wavelength: 0.97626 Å Processing
Processing