 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-6shh: Human kallikrein 7 with aromatic coumarinic ester compound 1 cova... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6shh | ||||||
|---|---|---|---|---|---|---|---|
| Title | Human kallikrein 7 with aromatic coumarinic ester compound 1 covalently bound to H57 | ||||||
 Components Components | Kallikrein-7 | ||||||
 Keywords Keywords | HYDROLASE / Serine Protease / Covalent Inhibitor / Complex | ||||||
| Function / homology |  Function and homology information Function and homology informationstratum corneum chymotryptic enzyme / epidermal lamellar body / antibacterial peptide biosynthetic process / cornified envelope / Differentiation of Keratinocytes in Interfollicular Epidermis in Mammalian Skin / extracellular matrix disassembly / epidermis development / Degradation of the extracellular matrix / secretory granule / serine-type peptidase activity ...stratum corneum chymotryptic enzyme / epidermal lamellar body / antibacterial peptide biosynthetic process / cornified envelope / Differentiation of Keratinocytes in Interfollicular Epidermis in Mammalian Skin / extracellular matrix disassembly / epidermis development / Degradation of the extracellular matrix / secretory granule / serine-type peptidase activity / protein maturation / metalloendopeptidase activity / peptidase activity / serine-type endopeptidase activity / proteolysis / : / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Hanke, S. / Straeter, N. | ||||||
| Funding support |  Germany, 1items Germany, 1items
| ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2020 Title: Structural Studies on the Inhibitory Binding Mode of Aromatic Coumarinic Esters to Human Kallikrein-Related Peptidase 7. Authors: Hanke, S. / Tindall, C.A. / Pippel, J. / Ulbricht, D. / Pirotte, B. / Reboud-Ravaux, M. / Heiker, J.T. / Strater, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6shh.cif.gz | 749.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6shh.ent.gz | 621.5 KB | Display | PDB format |
| PDBx/mmJSON format | 6shh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/sh/6shhftp://data.pdbj.org/pub/pdb/validation_reports/sh/6shh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  6shiC  6sjuC  6y4sC  2qxiS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj

- Assembly
Assembly









-Components
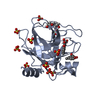
-Protein , 1 types, 8 molecules ABCDEFGH
| #1: Protein | Mass: 24481.160 Da / Num. of mol.: 8 Source method: isolated from a genetically manipulated source Details: The inhibitor is covalently attached to H57 / Source: (gene. exp.) Homo sapiens (human) / Gene: KLK7, PRSS6, SCCE / Production host:  References: UniProt: P49862, stratum corneum chymotryptic enzyme |
|---|
-Non-polymers , 5 types, 1246 molecules 

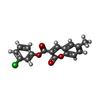


| #2: Chemical | ChemComp-PGE /  Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4 Mass: 150.173 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C6H14O4#3: Chemical | ChemComp-SO4 /  Mass: 96.063 Da / Num. of mol.: 83 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 83 / Source method: obtained synthetically / Formula: SO4#4: Chemical | ChemComp-SH7 / (  Mass: 314.720 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C17H11ClO4 Mass: 314.720 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: C17H11ClO4#5: Chemical |  Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1149 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Has ligand of interest | Y |
|---|---|
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.64 Å3/Da / Density % sol: 53.37 % |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop Details: 2.9 M ammonium sulphate, 0.1 M HEPES, pH 8.5, 0.5- 2 % PEG 3350 |
-Data collection
| Diffraction | Mean temperature: 100 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: BESSY / Beamline: 14.1 / Wavelength: 0.9184 Å |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Mar 28, 2018 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9184 Å / Relative weight: 1 |
| Reflection | Resolution: 2→48.49 Å / Num. obs: 138723 / % possible obs: 99.2 % / Redundancy: 9.4 % / Biso Wilson estimate: 30.04 Å2 / CC1/2: 0.993 / Rmerge(I) obs: 0.33 / Rpim(I) all: 0.166 / Rrim(I) all: 0.37 / Net I/σ(I): 5.8 |
| Reflection shell | Resolution: 2→2.04 Å / Redundancy: 8.5 % / Rmerge(I) obs: 4.121 / Mean I/σ(I) obs: 0.5 / Num. unique obs: 5745 / CC1/2: 0.229 / Rrim(I) all: 4.638 / % possible all: 84.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 2qxi Resolution: 2→47.03 Å / Cor.coef. Fo:Fc: 0.942 / Cor.coef. Fo:Fc free: 0.92 / SU R Cruickshank DPI: 0.17 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.181 / SU Rfree Blow DPI: 0.155 / SU Rfree Cruickshank DPI: 0.151
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 171.15 Å2 / Biso mean: 43.07 Å2 / Biso min: 18.46 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.31 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→47.03 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.02 Å / Rfactor Rfree error: 0 / Total num. of bins used: 50
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
