 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 6ow2 | ||||||
|---|---|---|---|---|---|---|---|
| Title | X-ray Structure of Polypeptide Deformylase | ||||||
 Components Components | Peptide deformylase | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE INHIBITOR / Inhibitor / complex / metal protein / enzyme / HYDROLASE-HYDROLASE INHIBITOR complex | ||||||
| Function / homology |  Function and homology information Function and homology informationpeptide deformylase / : / peptide deformylase activity / translation / metal ion binding Similarity search - Function | ||||||
| Biological species |   Streptococcus pneumoniae (bacteria) Streptococcus pneumoniae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å | ||||||
 Authors Authors | Campobasso, N. / Spletstoser, J. / Ward, P. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2019 Title: Discovery of piperazic acid peptide deformylase inhibitors with in vivo activity for respiratory tract and skin infections. Authors: Spletstoser, J.T. / Dreabit, J. / Knox, A.N. / Benowitz, A. / Campobasso, N. / Ward, P. / Cui, G. / Lewandowski, T. / McCloskey, L. / Aubart, K.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 6ow2.cif.gz | 65.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb6ow2.ent.gz | 38.6 KB | Display | PDB format |
| PDBx/mmJSON format | 6ow2.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ow/6ow2ftp://data.pdbj.org/pub/pdb/validation_reports/ow/6ow2 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 22589.854 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus pneumoniae (bacteria)Gene: defB, def, def_1, def_2, def_3, A5N45_09745, ERS019420_01538, ERS019688_00954, ERS020178_05158, ERS020534_01685, ERS020726_00048, ERS021057_00383, ERS021733_04069, ERS050646_01998, ERS367886_ ...Gene: defB, def, def_1, def_2, def_3, A5N45_09745, ERS019420_01538, ERS019688_00954, ERS020178_05158, ERS020534_01685, ERS020726_00048, ERS021057_00383, ERS021733_04069, ERS050646_01998, ERS367886_01588, ERS409327_03241, ERS409593_03947, KK0981_34260, NCTC12140_00964 Production host: References: UniProt: Q939R9, UniProt: Q8DP79*PLUS, peptide deformylase |
|---|---|
| #2: Chemical | ChemComp-NI /   Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni |
| #3: Chemical | ChemComp-NB4 / (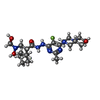  Mass: 481.564 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H36FN7O4 / Feature type: SUBJECT OF INVESTIGATION Mass: 481.564 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C22H36FN7O4 / Feature type: SUBJECT OF INVESTIGATION |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 86 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.71 Å3/Da / Density % sol: 54.59 % / Description: rods |
|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: 1 % - 4 % PEG400 40% - 56% Ammonium Sulfate 0.1 HEPES |
-Data collection
| Diffraction | Mean temperature: 175 K / Serial crystal experiment: N |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Nov 20, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→91 Å / Num. obs: 26688 / % possible obs: 88 % / Redundancy: 4.2 % / Biso Wilson estimate: 19.63 Å2 / Net I/σ(I): 18 |
| Reflection shell | Resolution: 1.6→1.63 Å / Num. unique obs: 1515 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.7→33.85 Å / SU ML: 0.2036 / Cross valid method: THROUGHOUT / σ(F): 1.36 / Phase error: 24.0664
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→33.85 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell |
|
