Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 13500.163 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
#2: Protein
Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 18302.613 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
#3: Protein
Dipeptidylpeptidase1 / Cathepsin C / Cathepsin J / Dipeptidyl peptidase I / DPPI / Dipeptidyl transferase
Mass: 7583.444 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: CTSC, CPPI / Cell line (production host): BTI-TN-5B1-4 / Production host: Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
Mass: 18.015 Da / Num. of mol.: 349 / Source method: isolated from a natural source / Formula: H2O
-
Details
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.79 Å3/Da / Density % sol: 55.94 %
Crystal grow
Temperature: 291 K / Method: vapor diffusion, sitting drop Details: 20.0 % (w/v) PEG 3000 0.2 M ammonium citrate dibasic 2.5 mM DTT
-
Data collection
Diffraction
Mean temperature: 100 K / Serial crystal experiment: N
Diffraction source
Source: ROTATING ANODE / Type: BRUKER X8 PROTEUM / Wavelength: 1.5418 Å
Detector
Type: Bruker Platinum 135 / Detector: CCD / Date: Dec 1, 2010
Radiation
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 1.5418 Å / Relative weight: 1
Reflection
Resolution: 2→28.3 Å / Num. obs: 30537 / % possible obs: 98.2 % / Redundancy: 11.5 % / Rmerge(I) obs: 0.069 / Net I/σ(I): 23.8
Reflection shell
Resolution: 2→2.1 Å / Rmerge(I) obs: 0.164 / Mean I/σ(I) obs: 6.1 / % possible all: 85.5
-
Processing
Software
Name
Version
Classification
REFMAC
5.8.02382018/15/10
refinement
PROTEUM
datareduction
PROTEUM
datascaling
REFMAC
phasing
Refinement
Method to determine structure: FOURIER SYNTHESIS / Resolution: 2→28.231 Å / Cor.coef. Fo:Fc: 0.961 / Cor.coef. Fo:Fc free: 0.94 / SU B: 4.767 / SU ML: 0.085 / Cross valid method: THROUGHOUT / ESU R: 0.155 / ESU R Free: 0.144 Details: Hydrogens have been added in their riding positions
Rfactor
Num. reflection
% reflection
Rfree
0.2034
1466
-
Rwork
0.1596
-
-
all
0.162
-
-
obs
-
29338
97.252 %
Solvent computation
Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å
Displacement parameters
Biso mean: 20.023 Å2
Baniso -1
Baniso -2
Baniso -3
1-
0.146 Å2
0 Å2
0 Å2
2-
-
0.417 Å2
0 Å2
3-
-
-
-0.563 Å2
Refinement step
Cycle: LAST / Resolution: 2→28.231 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
2759
0
127
349
3235
Refine LS restraints
Refine-ID
Type
Dev ideal
Dev ideal target
Number
X-RAY DIFFRACTION
r_bond_refined_d
0.014
0.013
3061
X-RAY DIFFRACTION
r_bond_other_d
0.002
0.018
2619
X-RAY DIFFRACTION
r_angle_refined_deg
1.931
1.709
4195
X-RAY DIFFRACTION
r_angle_other_deg
1.462
1.633
6113
X-RAY DIFFRACTION
r_dihedral_angle_1_deg
19.05
5.346
376
X-RAY DIFFRACTION
r_dihedral_angle_2_deg
29.536
22.933
150
X-RAY DIFFRACTION
r_dihedral_angle_3_deg
14.674
15
436
X-RAY DIFFRACTION
r_dihedral_angle_other_3_deg
4.877
15
3
X-RAY DIFFRACTION
r_dihedral_angle_4_deg
23.757
15
11
X-RAY DIFFRACTION
r_chiral_restr
0.091
0.2
403
X-RAY DIFFRACTION
r_gen_planes_refined
0.012
0.02
3705
X-RAY DIFFRACTION
r_gen_planes_other
0.004
0.02
691
X-RAY DIFFRACTION
r_nbd_refined
0.229
0.2
634
X-RAY DIFFRACTION
r_symmetry_nbd_other
0.19
0.2
2602
X-RAY DIFFRACTION
r_nbtor_refined
0.184
0.2
1489
X-RAY DIFFRACTION
r_symmetry_nbtor_other
0.093
0.2
1337
X-RAY DIFFRACTION
r_xyhbond_nbd_refined
0.212
0.2
297
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_other
0.093
0.2
1
X-RAY DIFFRACTION
r_symmetry_nbd_refined
0.244
0.2
19
X-RAY DIFFRACTION
r_nbd_other
0.216
0.2
67
X-RAY DIFFRACTION
r_symmetry_xyhbond_nbd_refined
0.33
0.2
22
X-RAY DIFFRACTION
r_mcbond_it
1.396
1.401
1424
X-RAY DIFFRACTION
r_mcbond_other
1.395
1.4
1423
X-RAY DIFFRACTION
r_mcangle_it
2.349
2.092
1786
X-RAY DIFFRACTION
r_mcangle_other
2.349
2.093
1787
X-RAY DIFFRACTION
r_scbond_it
1.358
1.574
1637
X-RAY DIFFRACTION
r_scbond_other
1.358
1.575
1638
X-RAY DIFFRACTION
r_scangle_it
2.225
2.318
2401
X-RAY DIFFRACTION
r_scangle_other
2.225
2.319
2402
X-RAY DIFFRACTION
r_lrange_it
5.599
18.088
3671
X-RAY DIFFRACTION
r_lrange_other
5.601
18.085
3671
LS refinement shell
Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 20
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Num. reflection all
% reflection obs (%)
WRfactor Rwork
2-2.052
0.202
75
0.185
1741
2165
83.8799
0.161
2.052-2.108
0.255
100
0.189
1862
2154
91.0863
0.165
2.108-2.168
0.246
104
0.175
1914
2085
96.7866
0.153
2.168-2.235
0.213
93
0.171
1906
2024
98.7648
0.148
2.235-2.307
0.218
103
0.159
1809
1934
98.8625
0.14
2.307-2.388
0.231
88
0.163
1782
1894
98.7328
0.144
2.388-2.477
0.229
105
0.164
1728
1849
99.1347
0.146
2.477-2.578
0.209
101
0.144
1671
1787
99.1606
0.132
2.578-2.691
0.16
79
0.149
1581
1672
99.2823
0.138
2.691-2.821
0.239
88
0.163
1564
1665
99.2192
0.156
2.821-2.972
0.209
72
0.155
1428
1512
99.2063
0.152
2.972-3.15
0.167
67
0.155
1408
1492
98.8606
0.155
3.15-3.365
0.22
65
0.165
1313
1388
99.2795
0.17
3.365-3.63
0.22
65
0.165
1227
1302
99.2319
0.175
3.63-3.97
0.161
67
0.139
1128
1209
98.842
0.153
3.97-4.428
0.167
49
0.131
1038
1094
99.3601
0.153
4.428-5.092
0.136
45
0.123
909
956
99.7908
0.151
5.092-6.186
0.22
44
0.176
804
849
99.8822
0.199
6.186-8.546
0.246
35
0.191
640
676
99.8521
0.226
8.546-28.231
0.266
21
0.239
408
431
99.536
0.305
Refinement TLS params.
Details: U values: with tls added / Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
1.6197
-0.0383
0.4764
1.2551
0.1648
1.2324
-0.0107
0.0025
0.0879
-0.0938
-0.0211
0.1128
-0.1275
-0.1008
0.0318
0.0337
0.0198
0.0013
0.0214
0.0175
0.0446
-6.84
-25.289
-20.185
2
1.1303
0.0273
-0.0689
1.2679
0.2074
0.8449
-0.0148
-0.0109
-0.0433
-0.0044
0.0004
-0.1286
-0.0478
0.1379
0.0145
0.0049
-0.0086
0.0006
0.0298
0.0161
0.0357
17.601
-30.779
-17.978
3
0.9605
0.0522
-0.1637
1.2573
0.3884
1.1713
0.019
-0.1569
0.0732
0.1278
0.0034
-0.1607
-0.1228
0.2362
-0.0223
0.0792
-0.0386
-0.0191
0.1131
0.0114
0.0707
20.239
-24.406
-8.854
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection
Auth asym-ID
Auth seq-ID
1
X-RAY DIFFRACTION
1
ALL
A
1 - 118
2
X-RAY DIFFRACTION
2
ALL
B
206 - 367
3
X-RAY DIFFRACTION
3
ALL
C
372 - 439
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Poland, 1items
Poland, 1items  Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads









 PDBj
PDBj










 Assembly
Assembly

 Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
Trichoplusia ni (cabbage looper) / References: UniProt: P53634, dipeptidyl-peptidase I
 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2
Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 2
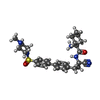

 Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl
Mass: 35.453 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Cl Mass: 521.674 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H35N5O3S / Feature type: SUBJECT OF INVESTIGATION
Mass: 521.674 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C28H35N5O3S / Feature type: SUBJECT OF INVESTIGATION Sample preparation
Sample preparation Processing
Processing