| Entry | Database: PDB / ID: 5my8
|
|---|
| Title | Crystal structure of SRPK1 in complex with SPHINX31 |
|---|
 Components Components | SRSF protein kinase 1,SRSF protein kinase 1 |
|---|
 Keywords Keywords | TRANSFERASE / splicing kinase / inhibitor / Structural Genomics / Structural Genomics Consortium / SGC |
|---|
| Function / homology |  Function and homology information Function and homology information
sperm DNA condensation / regulation of mRNA processing / regulation of mRNA splicing, via spliceosome / Maturation of nucleoprotein / negative regulation of viral genome replication / spliceosomal complex assembly / positive regulation of viral genome replication / Replacement of protamines by nucleosomes in the male pronucleus / RNA splicing / chromosome segregation ...sperm DNA condensation / regulation of mRNA processing / regulation of mRNA splicing, via spliceosome / Maturation of nucleoprotein / negative regulation of viral genome replication / spliceosomal complex assembly / positive regulation of viral genome replication / Replacement of protamines by nucleosomes in the male pronucleus / RNA splicing / chromosome segregation / nuclear matrix / protein phosphorylation / protein kinase activity / non-specific serine/threonine protein kinase / nuclear speck / intracellular signal transduction / innate immune response / protein serine kinase activity / protein serine/threonine kinase activity / chromatin / magnesium ion binding / RNA binding / nucleoplasm / ATP binding / nucleus / cytosol / cytoplasmSimilarity search - Function : / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site ...: / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha BetaSimilarity search - Domain/homology |
|---|
| Biological species |  Homo sapiens (human) Homo sapiens (human) |
|---|
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.7 Å |
|---|
 Authors Authors | Chaikuad, A. / von Delft, F. / Bountra, C. / Arrowsmith, C.H. / Edwards, A.M. / Knapp, S. / Structural Genomics Consortium (SGC) |
|---|
 Citation Citation | Journal: ACS Chem. Biol. / Year: 2017
Title: Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease.
Authors: Batson, J. / Toop, H.D. / Redondo, C. / Babaei-Jadidi, R. / Chaikuad, A. / Wearmouth, S.F. / Gibbons, B. / Allen, C. / Tallant, C. / Zhang, J. / Du, C. / Hancox, J.C. / Hawtrey, T. / Da ...Authors: Batson, J. / Toop, H.D. / Redondo, C. / Babaei-Jadidi, R. / Chaikuad, A. / Wearmouth, S.F. / Gibbons, B. / Allen, C. / Tallant, C. / Zhang, J. / Du, C. / Hancox, J.C. / Hawtrey, T. / Da Rocha, J. / Griffith, R. / Knapp, S. / Bates, D.O. / Morris, J.C. |
|---|
| History | | Deposition | Jan 25, 2017 | Deposition site: PDBE / Processing site: PDBE |
|---|
| Revision 1.0 | May 10, 2017 | Provider: repository / Type: Initial release |
|---|
| Revision 1.1 | May 8, 2024 | Group: Data collection / Database references / Category: chem_comp_atom / chem_comp_bond / database_2
Item: _database_2.pdbx_DOI / _database_2.pdbx_database_accession |
|---|
|
|---|
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads











 PDBj
PDBj Assembly
Assembly





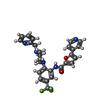

 Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7
Mass: 192.124 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H8O7 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM
Mass: 78.133 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6OS / Comment: DMSO, precipitant*YM Mass: 62.068 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C2H6O2
Mass: 62.068 Da / Num. of mol.: 20 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 507.507 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H24F3N5O2
Mass: 507.507 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H24F3N5O2 Sample preparation
Sample preparation / Beamline: I02 / Wavelength: 0.97949 Å
/ Beamline: I02 / Wavelength: 0.97949 Å Processing
Processing