 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5mdh: CRYSTAL STRUCTURE OF TERNARY COMPLEX OF PORCINE CYTOPLASMIC MALAT... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5mdh | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TERNARY COMPLEX OF PORCINE CYTOPLASMIC MALATE DEHYDROGENASE ALPHA-KETOMALONATE AND TNAD AT 2.4 ANGSTROMS RESOLUTION | ||||||
 Components Components | MALATE DEHYDROGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / (NAD(A)-CHOH(D)) | ||||||
| Function / homology |  Function and homology information Function and homology information(2R)-hydroxyphenylpyruvate reductase [NAD(P)H] activity / Malate-aspartate shuttle / (S)-malate dehydrogenase (NAD+, oxaloacetate-forming) / L-malate dehydrogenase (NAD+) activity / malate metabolic process / oxaloacetate metabolic process / NAD+ metabolic process / tricarboxylic acid cycle / NAD binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Chapman, A.D.M. / Cortes, A. / Dafforn, T.R. / Clarke, A.R. / Brady, R.L. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1999 Title: Structural basis of substrate specificity in malate dehydrogenases: crystal structure of a ternary complex of porcine cytoplasmic malate dehydrogenase, alpha-ketomalonate and tetrahydoNAD. Authors: Chapman, A.D. / Cortes, A. / Dafforn, T.R. / Clarke, A.R. / Brady, R.L. #1: Journal: Biochemistry / Year: 1989Title: Refined Crystal Structure of Cytoplasmic Malate Dehydrogenase at 2.5-A Resolution Authors: Birktoft, J.J. / Rhodes, G. / Banaszak, L.J. #2: Journal: Biochemistry / Year: 1987Title: Structure of Porcine Heart Cytoplasmic Malate Dehydrogenase. Combining X-Ray Diffraction and Chemical Sequence Data in Structural Studies Authors: Birktoft, J.J. / Bradshaw, R.A. / Banaszak, L.J. #3: Journal: J.Biol.Chem. / Year: 1983Title: The Presence of a Histidine-Aspartic Acid Pair in the Active Site of 2-Hydroxyacid Dehydrogenases. X-Ray Refinement of Cytoplasmic Malate Dehydrogenase Authors: Birktoft, J.J. / Banaszak, L.J. #4: Journal: MOLECULAR STRUCTURE AND BIOLOGICAL ACTIVITY / Year: 1982Title: The Interactions of Nad/Nadh with 2-Hydroxy Acid Dehydrogenases Authors: Birktoft, J.J. / Fernley, R.T. / Bradshaw, R.A. / Banaszak, L.J. #5: Journal: Proc.Natl.Acad.Sci.USA / Year: 1982Title: Amino Acid Sequence Homology Among the 2-Hydroxy Acid Dehydrogenases. Mitochondrial and Cytoplasmic Malate Dehydrogenases Form a Homologous System with Lactate Dehydrogenase Authors: Birktoft, J.J. / Fernley, R.T. / Bradshaw, R.A. / Banaszak, L.J. #6: Journal: STRUCTURE AND CONFORMATION OF NUCLEIC ACIDS AND PROTEIN-NUCLEIC ACID INTERACTIONS : PROCEEDINGS OF THE FOURTH ANNUAL HARRY STEENBOCK SYMPOSIUM, JUNE 16-19, 1974, MADISON, WISCONSINYear: 1975 Title: Nicotinamide Adenine Dinucleotide and the Active Site of Cytoplasmic Malate Dehydrogenase Authors: Banaszak, L.J. / Webb, L.E. #7: Journal: Biochemistry / Year: 1973Title: Conformation of Nicotinamide Adenine Dinucleotide Bound to Cytoplasmic Malate Dehydrogenase Authors: Webb, L.E. / Hill, E.J. / Banaszak, L.J. #8: Journal: J.Mol.Biol. / Year: 1972Title: Polypeptide Conformation of Cytoplasmic Malate Dehydrogenase from an Electron Density Map at 3.0 Angstroms Resolution Authors: Hill, E. / Tsernoglou, D. / Webb, L. / Banaszak, L.J. #9: Journal: Biochem.Biophys.Res.Commun. / Year: 1972Title: The Identification of an Asymmetric Complex of Nicotinamide Adenine Dinucleotide and Pig Heart Cytoplasmic Malate Dehydrogenase Authors: Glatthaar, B.E. / Banaszak, L.J. / Bradshaw, R.A. #10: Journal: J.Mol.Biol. / Year: 1972Title: Cytoplasmic Malate Dehydrogenase--Heavy Atom Derivatives and Low Resolution Structure Authors: Tsernoglou, D. / Hill, E. / Banaszak, L.J. #11: Journal: Cold Spring Harbor Symp.Quant.Biol. / Year: 1972Title: Structural Studies on Heart Muscle Malate Dehydrogenases Authors: Tsernoglou, D. / Hill, E. / Banaszak, L.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5mdh.cif.gz | 148.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5mdh.ent.gz | 117.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5mdh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/md/5mdhftp://data.pdbj.org/pub/pdb/validation_reports/md/5mdh | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  4mdhS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
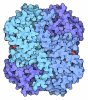

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | THE ASYMMETRIC UNIT CONTAINS TWO SUBUNITS WHICH HAVE BEEN ASSIGNED CHAIN IDENTIFIERS *A* AND *B*. ALTHOUGH THE TWO SUBUNITS ARE CHEMICALLY EQUIVALENT, THEY ARE RELATED BY A NON-CRYSTALLOGRAPHIC SYMMETRY AXIS WITH A ROTATION ANGLE OF 174.9 DEGREES WHICH IS SIGNIFICANTLY DIFFERENT FROM A TRUE TWO-FOLD SYMMETRY OPERATION. |
-Components
| #1: Protein | Mass: 36367.941 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  References: UniProt: P11708, (S)-malate dehydrogenase (NAD+, oxaloacetate-forming) #2: Chemical |   Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM#3: Chemical | 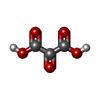  Mass: 118.045 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H2O5 Mass: 118.045 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H2O5#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 354 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.78 Å3/Da / Density % sol: 57.1 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.6 / Details: pH 5.6 | |||||||||||||||
| Crystal | *PLUS | |||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.488 / Beamline: PX7.2 / Wavelength: 1.488 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 1, 1996 / Details: MIRRORS |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→15 Å / Num. obs: 26268 / % possible obs: 89.3 % / Observed criterion σ(I): 0 / Redundancy: 3.96 % / Biso Wilson estimate: 42.44 Å2 / Rmerge(I) obs: 0.093 / Rsym value: 0.093 / Net I/σ(I): 12.34 |
| Reflection shell | Resolution: 2.4→2.51 Å / Redundancy: 3.9 % / Rmerge(I) obs: 0.32 / Mean I/σ(I) obs: 3.72 / Rsym value: 0.32 / % possible all: 91.6 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 4MDH Resolution: 2.4→15 Å / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.67 / ESU R Free: 0.3 Details: ATOMS WITH ZERO OCCUPANCIES HAVE NO ELECTRON DENSITY IN THE MAPS, THEREFORE THE POSITIONS OF THESE RESIDUES CANNOT BE DETERMINED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 46.38 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→15 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.199 / Rfactor Rfree: 0.25378 / Rfactor Rwork: 0.19946 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
