 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-5f03: TRYPTASE B2 IN COMPLEX WITH 5-(3-Aminomethyl-phenoxymethyl)-3-[3-... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5f03 | ||||||
|---|---|---|---|---|---|---|---|
| Title | TRYPTASE B2 IN COMPLEX WITH 5-(3-Aminomethyl-phenoxymethyl)-3-[3-(2-chloro-pyridin-3-ylethynyl)-phenyl]-oxazolidin-2-one; compound with trifluoro-acetic acid | ||||||
 Components Components | Tryptase beta-2 | ||||||
 Keywords Keywords | HYDROLASE / HUMAN TRYPTASE / SERINE PROTEINASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtryptase / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / serine-type peptidase activity / defense response / extracellular matrix / serine-type endopeptidase activity / proteolysis / : / extracellular region / identical protein binding Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.94 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.94 Å | ||||||
 Authors Authors | Banner, D. / Benz, J. / Joseph, C. / Kuglstatter, A. | ||||||
 Citation Citation | Journal: J.Med.Chem. / Year: 2016 Title: A Real-World Perspective on Molecular Design. Authors: Kuhn, B. / Guba, W. / Hert, J. / Banner, D. / Bissantz, C. / Ceccarelli, S. / Haap, W. / Korner, M. / Kuglstatter, A. / Lerner, C. / Mattei, P. / Neidhart, W. / Pinard, E. / Rudolph, M.G. / ...Authors: Kuhn, B. / Guba, W. / Hert, J. / Banner, D. / Bissantz, C. / Ceccarelli, S. / Haap, W. / Korner, M. / Kuglstatter, A. / Lerner, C. / Mattei, P. / Neidhart, W. / Pinard, E. / Rudolph, M.G. / Schulz-Gasch, T. / Woltering, T. / Stahl, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5f03.cif.gz | 222.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5f03.ent.gz | 176.5 KB | Display | PDB format |
| PDBx/mmJSON format | 5f03.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f0/5f03ftp://data.pdbj.org/pub/pdb/validation_reports/f0/5f03 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5edbC  5edcC  5edeC  5edgC  5edhC  5ediC  5ezxC  5ezzC  5f00C  5f01C  5f02C  5i2rC C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27491.426 Da / Num. of mol.: 2 / Mutation: NO Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: MAST CELLS / Gene: TPSB2, TPS2 / Production host:  Komagataella pastoris (fungus) / References: UniProt: P20231, UniProt: Q15661*PLUS, tryptase Komagataella pastoris (fungus) / References: UniProt: P20231, UniProt: Q15661*PLUS, tryptase#2: Chemical | 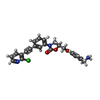  Mass: 433.887 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H20ClN3O3 Mass: 433.887 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C24H20ClN3O3#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 689 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 689 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.67 Å3/Da / Density % sol: 53.99 % |
|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 7.5 / Details: 20% PEG 10000, 0.1 M HEPES |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 Å |
| Detector | Type: MAR scanner 345 mm plate / Detector: IMAGE PLATE / Date: Dec 1, 2006 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.94→67.88 Å / Num. obs: 43956 / % possible obs: 98.8 % / Redundancy: 5.4 % / Rmerge(I) obs: 0.096 / Net I/av σ(I): 13.84 / Net I/σ(I): 0.0103 |
| Reflection shell | Resolution: 1.94→2.06 Å / Redundancy: 5.2 % / Rmerge(I) obs: 0.249 / Mean I/σ(I) obs: 4.78 / % possible all: 93.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: in-house structure Resolution: 1.94→67.88 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.935 / SU B: 5.787 / SU ML: 0.089 / Cross valid method: THROUGHOUT / ESU R: 0.139 / ESU R Free: 0.133 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 15.628 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.94→67.88 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|