 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-5301: Negative Stain reconstruction of the Thermus thermophilus A-ATPas... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-5301 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Negative Stain reconstruction of the Thermus thermophilus A-ATPase to 23 Angstrom. Opposite Hand to published. | |||||||||
 Map data Map data | This is a negative stain reconstruction of the Thermus thermophilus A-ATPase | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | A-ATPase / Thermus thermophilus / ATPase | |||||||||
| Biological species |   Thermus thermophilus (bacteria) Thermus thermophilus (bacteria) | |||||||||
| Method | single particle reconstruction / negative staining / Resolution: 23.0 Å | |||||||||
 Authors Authors | Bernal RA / Stock D | |||||||||
 Citation Citation | Journal: Structure / Year: 2004 Title: Three-dimensional structure of the intact Thermus thermophilus H+-ATPase/synthase by electron microscopy. Authors: Ricardo A Bernal / Daniela Stock /  Abstract: ATPases are unique rotary motors that are essential to all living organisms because of their role in energy interconversion. A three-dimensional reconstruction of the intact H+-ATPase/synthase from ...ATPases are unique rotary motors that are essential to all living organisms because of their role in energy interconversion. A three-dimensional reconstruction of the intact H+-ATPase/synthase from Thermus thermophilus has revealed the presence of two interconnected peripheral stalks, a well-defined central stalk, and a hexagonally shaped hydrophobic domain. The peripheral stalks are each attached to the water soluble sector at a noncatalytic subunit interface and extend down toward the membrane where they interact with a strong elongated tube of density that runs parallel to the membrane and connects the two stalks. The central stalk is well resolved, especially with respect to its interaction with a single catalytic subunit giving rise to an asymmetry comparable to that identified in F-ATPases. The hexagonal shape of the membrane domain might suggest the presence of 12 proteolipids arranged as dimers, analogous to the proposed arrangement in the related eukaryotic V-ATPases. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
 UCSF Chimera
UCSF Chimera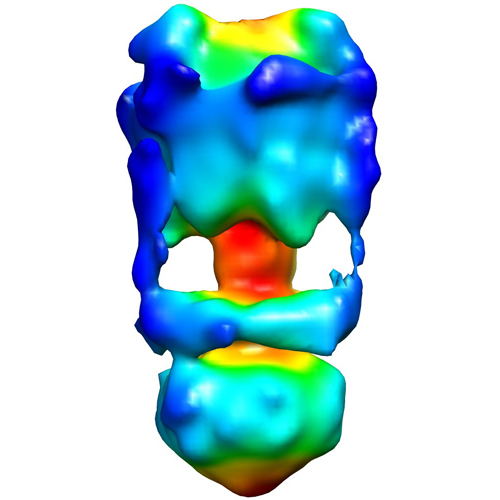
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_5301.map.gz | 7.5 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-5301-v30.xmlemd-5301.xml | 14.3 KB 14.3 KB | Display Display | EMDB header |
| Images | 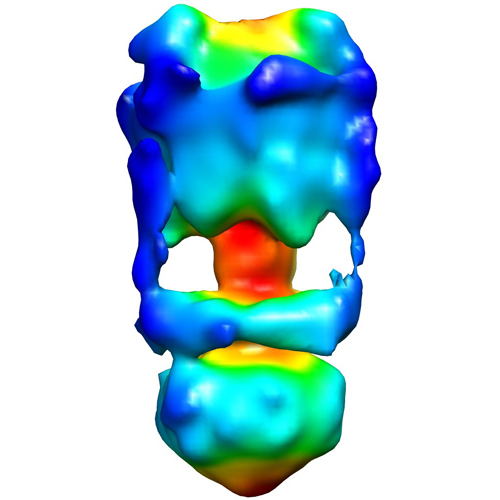 emd_5301_1.jpg emd_5301_1.jpg | 118.2 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5301ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5301 http://ftp.pdbj.org/pub/emdb/structures/EMD-5301ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5301 | HTTPS FTP |
-Related structure data
| Similar structure data |
|---|







-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_5301.map.gz / Format: CCP4 / Size: 7.8 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | This is a negative stain reconstruction of the Thermus thermophilus A-ATPase | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 3.3 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Thermus thermophilus A-ATPase
| Entire | Name: Thermus thermophilus A-ATPase |
|---|---|
| Components |
|
-Supramolecule #1000: Thermus thermophilus A-ATPase
| Supramolecule | Name: Thermus thermophilus A-ATPase / type: sample / ID: 1000 / Oligomeric state: multi-subunit complex / Number unique components: 1 |
|---|
-Supramolecule #1: ATPase
| Supramolecule | Name: ATPase / type: organelle_or_cellular_component / ID: 1 / Name.synonym: ATPase / Oligomeric state: multimer / Recombinant expression: No / Database: NCBI |
|---|---|
| Source (natural) | Organism: Thermus thermophilus (bacteria) / Strain: HB8 / synonym: Thermus thermophilus / Location in cell: membrane |
-Experimental details
-Structure determination
| Method | negative staining |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.02 mg/mL |
|---|---|
| Buffer | pH: 8 Details: 20 mM Tris, pH 8.0, 2 mM MgCl2, 1 mM EDTA, 0.05% n-dodecyl-beta-D-maltoside, 0.02% NaN3 |
| Staining | Type: NEGATIVE Details: Three microliters of the T. thermophilus sample, diluted to 0.02 mg/mL, was placed onto the surface of the carbon-coated grid. The sample was blotted off and replaced with 3 microliters of ...Details: Three microliters of the T. thermophilus sample, diluted to 0.02 mg/mL, was placed onto the surface of the carbon-coated grid. The sample was blotted off and replaced with 3 microliters of 2% uranyl acetate. The uranyl acetate was blotted away and replaced with 3 microliters of 4% methylamine tungstate. The final drop of methylamine tungstate was blotted away and the grid was left to air dry. |
| Grid | Details: 400 mesh 3.05 mm copper grids with a thin layer carbon support |
| Vitrification | Cryogen name: NONE / Instrument: OTHER |
- Electron microscopy
Electron microscopy
| Microscope | FEI TECNAI 12 |
|---|---|
| Temperature | Average: 25 K |
| Alignment procedure | Legacy - Astigmatism: not corrected |
| Date | Jan 22, 2003 |
| Image recording | Category: FILM / Film or detector model: KODAK SO-163 FILM / Digitization - Scanner: ZEISS SCAI / Digitization - Sampling interval: 14 µm / Bits/pixel: 8 |
| Electron beam | Acceleration voltage: 120 kV / Electron source: LAB6 |
| Electron optics | Calibrated magnification: 42000 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 1.0 µm / Nominal defocus min: 1.0 µm / Nominal magnification: 42000 |
| Sample stage | Specimen holder: side entry single tilt / Specimen holder model: OTHER |
-Image processing
| CTF correction | Details: no ctf correction done because it was a negative stain reconstruction |
|---|---|
| Final reconstruction | Algorithm: OTHER / Resolution.type: BY AUTHOR / Resolution: 23.0 Å / Resolution method: FSC 0.5 CUT-OFF / Software - Name: MRC Image2000 and Imagic / Number images used: 12300 |
| Final two d classification | Number classes: 60 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F / Chain - #6 - Chain ID: G |
|---|---|
| Software | Name: EMfit |
| Details | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: sumf and number of atoms inside density |

-Atomic model buiding 2
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C |
|---|---|
| Software | Name: EMfit |
| Details | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: sumf and number of atoms inside density |

-Atomic model buiding 3
| Initial model | PDB ID: Chain - #0 - Chain ID: A / Chain - #1 - Chain ID: B / Chain - #2 - Chain ID: C / Chain - #3 - Chain ID: D / Chain - #4 - Chain ID: E / Chain - #5 - Chain ID: F / Chain - #6 - Chain ID: G / Chain - #7 - Chain ID: H / Chain - #8 - Chain ID: I / Chain - #9 - Chain ID: J / Chain - #10 - Chain ID: K / Chain - #11 - Chain ID: L |
|---|---|
| Software | Name: EMfit |
| Details | PDBEntryID_givenInChain. Protocol: Each chain was fit as a separate rigid body. X-ray coordinates for the bovine alpha3 beta3 and gamma sub-assemblies were manually fitted into the EM density using the program O. The program EMfit (Rossmann et al., 2001) was then used in order to obtain a more quantitative fit. |
| Refinement | Space: REAL / Protocol: RIGID BODY FIT / Target criteria: sumf and number of atoms inside density |

