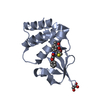
- PDB-4ibe: Ebola virus VP35 bound to small molecule -
+
Open data
ID or keywords:
Loading...
-
Basic information
Entry
Database: PDB / ID: 4ibe
Title
Ebola virus VP35 bound to small molecule
Components
Polymerase cofactor VP35
Keywords
TRANSCRIPTION/TRANSCRIPTION inhibitor / Structural Genomics / NIAID / National Institute of Allergy and Infectious Diseases / Center for Structural Genomics of Infectious Diseases / CSGID / interferon inhibitor domain / TRANSCRIPTION-TRANSCRIPTION inhibitor complex
Function / homology
Function and homology information
symbiont-mediated suppression of host immune response / symbiont-mediated suppression of host RNAi-mediated antiviral immune response / negative regulation of miRNA-mediated gene silencing / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of IKBKE activity / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of IRF7 activity / symbiont-mediated suppression of host antigen processing and presentation of peptide antigen via MHC class II / symbiont-mediated suppression of host PKR/eIFalpha signaling / positive regulation of protein sumoylation / viral transcription / molecular sequestering activity ...symbiont-mediated suppression of host immune response / symbiont-mediated suppression of host RNAi-mediated antiviral immune response / negative regulation of miRNA-mediated gene silencing / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of IKBKE activity / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of IRF7 activity / symbiont-mediated suppression of host antigen processing and presentation of peptide antigen via MHC class II / symbiont-mediated suppression of host PKR/eIFalpha signaling / positive regulation of protein sumoylation / viral transcription / molecular sequestering activity / viral genome replication / viral nucleocapsid / symbiont-mediated suppression of host cytoplasmic pattern recognition receptor signaling pathway via inhibition of TBK1 activity / symbiont-mediated suppression of host toll-like receptor signaling pathway / host cell cytoplasm / symbiont-mediated suppression of host innate immune response / symbiont-mediated suppression of host type I interferon-mediated signaling pathway / negative regulation of gene expression / RNA binding Similarity search - Function
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray
Radiation wavelength
Wavelength: 0.979237 Å / Relative weight: 1
Reflection
Resolution: 1.95→50 Å / Num. obs: 17475 / % possible obs: 93.6 % / Redundancy: 4.4 % / Rmerge(I) obs: 0.115 / Net I/σ(I): 6
Reflection shell
Resolution (Å)
Redundancy (%)
Rmerge(I) obs
Diffraction-ID
% possible all
1.95-1.98
3.3
0.862
1
94.5
1.98-2.02
3.5
0.776
1
95.1
2.02-2.06
3.7
0.673
1
96.5
2.06-2.1
3.9
0.582
1
95.8
2.1-2.15
4.2
0.538
1
95.1
2.15-2.2
4.3
0.491
1
96.1
2.2-2.25
4.4
0.431
1
95.2
2.25-2.31
4.5
0.386
1
94.8
2.31-2.38
4.6
0.334
1
94.5
2.38-2.46
4.6
0.285
1
94.7
2.46-2.54
4.6
0.251
1
93.8
2.54-2.65
4.6
0.202
1
94.1
2.65-2.77
4.6
0.176
1
93.6
2.77-2.91
4.7
0.137
1
92.6
2.91-3.1
4.7
0.119
1
93.7
3.1-3.33
4.7
0.095
1
92.7
3.33-3.67
4.8
0.074
1
92.2
3.67-4.2
4.8
0.058
1
91.7
4.2-5.29
4.7
0.052
1
90.6
5.29-50
4.9
0.052
1
86
-
Phasing
Phasing
Method: molecular replacement
-
Processing
Software
Name
Version
Classification
NB
PHENIX
1.8.1_1168
refinement
SCALEPACK
datascaling
MOLREP
phasing
REFMAC
refinement
PDB_EXTRACT
3.11
dataextraction
HKL-3000
datacollection
DENZO
datareduction
Refinement
Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.95→48.502 Å / Cor.coef. Fo:Fc: 0.958 / Cor.coef. Fo:Fc free: 0.932 / Occupancy max: 1 / Occupancy min: 0.5 / SU ML: 0.16 / SU R Cruickshank DPI: 0.2719 / σ(F): 1.36 / Phase error: 28.15 / Stereochemistry target values: ML / Details: HYDROGENS HAVE BEEN USED IF PRESENT IN THE INPUT
Rfactor
Num. reflection
% reflection
Rfree
0.2326
1498
9.93 %
Rwork
0.1825
-
-
obs
0.1874
15087
80.23 %
Solvent computation
Shrinkage radii: 0.9 Å / VDW probe radii: 1.11 Å / Solvent model: FLAT BULK SOLVENT MODEL
Displacement parameters
Biso mean: 26.7313 Å2
Baniso -1
Baniso -2
Baniso -3
1-
-0.01 Å2
0 Å2
0 Å2
2-
-
0 Å2
0 Å2
3-
-
-
0.01 Å2
Refinement step
Cycle: LAST / Resolution: 1.95→48.502 Å
Protein
Nucleic acid
Ligand
Solvent
Total
Num. atoms
1917
0
68
163
2148
Refine LS restraints
Refine-ID
Type
Dev ideal
Number
X-RAY DIFFRACTION
f_bond_d
0.006
2085
X-RAY DIFFRACTION
f_angle_d
1.2
2843
X-RAY DIFFRACTION
f_dihedral_angle_d
16.467
821
X-RAY DIFFRACTION
f_chiral_restr
0.066
306
X-RAY DIFFRACTION
f_plane_restr
0.006
376
LS refinement shell
Resolution (Å)
Rfactor Rfree
Num. reflection Rfree
Rfactor Rwork
Num. reflection Rwork
Refine-ID
% reflection obs (%)
1.9358-1.9983
0.2765
81
0.205
596
X-RAY DIFFRACTION
40
1.9983-2.0697
0.264
77
0.2062
836
X-RAY DIFFRACTION
55
2.0697-2.1526
0.2148
115
0.198
1022
X-RAY DIFFRACTION
68
2.1526-2.2505
0.2638
134
0.2017
1176
X-RAY DIFFRACTION
77
2.2505-2.3692
0.2389
141
0.1947
1315
X-RAY DIFFRACTION
87
2.3692-2.5176
0.2855
142
0.1969
1434
X-RAY DIFFRACTION
93
2.5176-2.712
0.24
145
0.2019
1448
X-RAY DIFFRACTION
94
2.712-2.9849
0.2472
177
0.1884
1426
X-RAY DIFFRACTION
93
2.9849-3.4167
0.2204
144
0.1722
1445
X-RAY DIFFRACTION
93
3.4167-4.3043
0.1989
175
0.1532
1431
X-RAY DIFFRACTION
92
4.3043-48.5175
0.2383
167
0.1866
1460
X-RAY DIFFRACTION
89
Refinement TLS params.
Method: refined / Refine-ID: X-RAY DIFFRACTION
ID
L11 (°2)
L12 (°2)
L13 (°2)
L22 (°2)
L23 (°2)
L33 (°2)
S11 (Å °)
S12 (Å °)
S13 (Å °)
S21 (Å °)
S22 (Å °)
S23 (Å °)
S31 (Å °)
S32 (Å °)
S33 (Å °)
T11 (Å2)
T12 (Å2)
T13 (Å2)
T22 (Å2)
T23 (Å2)
T33 (Å2)
Origin x (Å)
Origin y (Å)
Origin z (Å)
1
3.4063
2.9993
-2.5386
7.4819
-0.986
2.704
-0.4314
0.753
-0.9429
-0.4743
0.297
0.2742
0.4408
-0.1959
0.015
0.2629
-0.0057
-0.0253
0.2471
-0.0528
0.2628
1.9543
22.5283
-12.2789
2
6.1445
-1.5183
0.0556
0.9682
-0.2983
2.5041
0.0486
0.2569
-0.1697
0.055
0.0186
0.1018
-0.171
-0.0252
-0.0224
0.115
-0.0238
0.0061
0.1223
-0.0261
0.1029
3.2349
29.1187
-8.4005
3
5.1565
1.4843
-4.7451
2.232
-0.7289
5.1358
0.1654
0.6035
0.7362
-0.2038
0.2557
0.2516
-0.5567
0.0138
-0.4167
0.1786
-0.0379
-0.0079
0.2107
0.04
0.2322
7.966
36.8719
-10.0172
4
9.4241
-5.2123
1.2148
7.8159
-2.7018
5.0076
-0.1507
-0.5254
-0.2595
0.6936
0.0952
0.1247
0.2865
-0.0635
0.0416
0.2674
-0.1033
0.0456
0.2631
-0.0298
0.172
5.0507
27.8885
5.2617
5
5.1575
2.0724
2.4132
3.5628
-0.141
6.7759
0.0049
-0.0774
0.3458
0.6647
-0.0991
-0.1319
0.3094
0.1818
0.1519
0.2228
-0.0242
-0.0048
0.2093
0.0104
0.1168
14.0871
31.0686
7.0997
6
5.7959
-1.5917
2.1795
8.7246
-5.396
3.9413
0.0627
-0.3574
0.0471
0.8352
0.1214
0.3761
-0.2045
-0.4975
-0.1753
0.2818
-0.0501
0.0525
0.2713
-0.0658
0.1383
9.2789
30.0227
8.4818
7
6.5595
-4.6543
0.8015
6.3978
-0.4532
5.584
0.1898
0.4256
-0.2125
0.1614
-0.2541
-0.1927
0.3864
0.1488
0.0749
0.2356
0.0291
-0.0379
0.1954
0.0502
0.2408
-1.8081
21.782
34.8772
8
6.4959
0.5706
-0.0056
1.5787
1.4491
4.3647
-0.0172
0.0522
-0.3084
-0.0271
0.0591
-0.109
-0.0151
0.1618
-0.0356
0.1144
0.0225
-0.0046
0.0662
0.0402
0.1355
-3.2336
28.3517
31.1043
9
6.8466
0.1035
-3.6599
0.8979
0.3172
6.1118
0.1127
-0.5938
0.4853
0.0325
0.0544
-0.1552
-0.3017
0.2328
-0.2147
0.1188
0.0277
0.0138
0.0956
-0.0244
0.2098
-4.6866
37.1473
30.0396
10
5.6802
3.1479
3.2337
7.9553
4.3897
4.9175
-0.3435
1.0584
-0.0654
-0.3573
-0.0877
0.0224
0.0059
0.1497
0.0708
0.2611
-0.0183
0.0344
0.1746
-0.1183
0.1519
-9.2412
20.8706
14.1439
11
4.614
-5.5753
1.233
8.4732
2.3834
8.9168
0.24
-0.5885
-0.3569
0.3848
-0.2239
1.2419
0.936
-0.6382
0.0816
0.1978
-0.0835
0.01
0.3833
0.0265
0.2965
-19.0108
24.1335
20.2294
12
5.9145
5.9944
1.6667
8.3326
0.2657
2.0833
0.0523
-0.2042
0.581
0.0487
-0.1208
0.3299
-0.6238
0.1714
0.2187
0.2325
-0.0098
0.0076
0.1897
0.0109
0.2691
-10.2825
36.1679
12.8272
13
6.495
0.5882
1.0326
5.0467
4.171
3.555
0.1351
0.1922
0.0798
-0.42
0.0421
-0.1818
-0.3588
-0.1463
-0.2529
0.2237
0.005
0.0226
0.1657
0.0553
0.1667
-9.2691
29.9328
14.6309
Refinement TLS group
ID
Refine-ID
Refine TLS-ID
Selection details
1
X-RAY DIFFRACTION
1
chain 'A' and (resid218through231 )
2
X-RAY DIFFRACTION
2
chain 'A' and (resid232through269 )
3
X-RAY DIFFRACTION
3
chain 'A' and (resid270through283 )
4
X-RAY DIFFRACTION
4
chain 'A' and (resid284through304 )
5
X-RAY DIFFRACTION
5
chain 'A' and (resid305through319 )
6
X-RAY DIFFRACTION
6
chain 'A' and (resid320through340 )
7
X-RAY DIFFRACTION
7
chain 'B' and (resid217through231 )
8
X-RAY DIFFRACTION
8
chain 'B' and (resid232through268 )
9
X-RAY DIFFRACTION
9
chain 'B' and (resid269through292 )
10
X-RAY DIFFRACTION
10
chain 'B' and (resid293through304 )
11
X-RAY DIFFRACTION
11
chain 'B' and (resid305through310 )
12
X-RAY DIFFRACTION
12
chain 'B' and (resid311through319 )
13
X-RAY DIFFRACTION
13
chain 'B' and (resid320through340 )
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information
 Ebola virus
Ebola virus X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj
 Assembly
Assembly



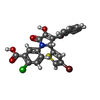
 Mass: 518.764 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H13BrClNO5S
Mass: 518.764 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C22H13BrClNO5S
 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3
Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 18.015 Da / Num. of mol.: 163 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 163 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: 19-ID / Wavelength: 0.979237 Å
/ Beamline: 19-ID / Wavelength: 0.979237 Å Processing
Processing