 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-3zdv: Crystal structure of the LecB lectin from Pseudomonas aeruginosa ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 3zdv | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of the LecB lectin from Pseudomonas aeruginosa in complex with Methyl 6-(2,4,6-trimethylphenylsulfonylamido)-6-deoxy-alpha-D-mannopyranoside | ||||||
 Components Components | FUCOSE-BINDING LECTIN PA-IIL | ||||||
 Keywords Keywords | SUGAR BINDING PROTEIN / GLYCOMIMETIC / INHIBITOR | ||||||
| Function / homology |  Function and homology information Function and homology informationsingle-species biofilm formation / carbohydrate binding / metal ion binding Similarity search - Function | ||||||
| Biological species |   PSEUDOMONAS AERUGINOSA (bacteria) PSEUDOMONAS AERUGINOSA (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.41 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.41 Å | ||||||
 Authors Authors | Hauck, D. / Joachim, I. / Frommeyer, B. / Varrot, A. / Philipp, B. / MOller, H.M. / Imberty, A. / Exner, T.E. / Titz, A. | ||||||
 Citation Citation | Journal: Acs Chem.Biol. / Year: 2013 Title: Discovery of Two Classes of Potent Glycomimetic Inhibitors of Pseudomonas Aeruginosa Lecb with Distinct Binding Modes. Authors: Hauck, D. / Joachim, I. / Frommeyer, B. / Varrot, A. / Philipp, B. / Moller, H.M. / Imberty, A. / Exner, T.E. / Titz, A. | ||||||
| History |
| ||||||
| Remark 700 | SHEET DETERMINATION METHOD: AUTHOR PROVIDED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 3zdv.cif.gz | 205.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb3zdv.ent.gz | 163.3 KB | Display | PDB format |
| PDBx/mmJSON format | 3zdv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/zd/3zdvftp://data.pdbj.org/pub/pdb/validation_reports/zd/3zdv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  3dcqS S: Starting model for refinement |
|---|---|
| Similar structure data |









-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||
| Unit cell |
| ||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
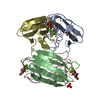
-Protein / Sugars , 2 types, 8 molecules ABCD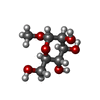
| #1: Protein | Mass: 11865.905 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) PSEUDOMONAS AERUGINOSA (bacteria) / Strain: PAO1 / Plasmid: PET25PA2 / Production host: #3: Sugar | ChemComp-MMA /  Type: D-saccharide / Mass: 194.182 Da / Num. of mol.: 4 Type: D-saccharide / Mass: 194.182 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C7H14O6 |
|---|
-Non-polymers , 6 types, 468 molecules 
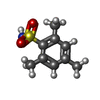




| #2: Chemical | ChemComp-CA /  Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 8 / Source method: obtained synthetically / Formula: Ca#4: Chemical | ChemComp-F1A /  Mass: 199.270 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H13NO2S Mass: 199.270 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H13NO2S#5: Chemical | ChemComp-GOL / |  Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#6: Chemical | ChemComp-EDO / |  Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2 Mass: 62.068 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C2H6O2#7: Chemical | ChemComp-SO4 / |  Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#8: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 453 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Details
| Sequence details | THE N-TERMINAL METHIONINE |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.89 Å3/Da / Density % sol: 34.93 % / Description: NONE |
|---|---|
| Crystal grow | pH: 6.5 Details: 330 MM SODIUM CACODYLATE PH 6.5 200 MM AMMONIUM SULPHATE 30% PEG8K |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SOLEIL  / Beamline: PROXIMA 1 / Wavelength: 0.97857 / Beamline: PROXIMA 1 / Wavelength: 0.97857 |
| Detector | Type: DECTRIS PILATUS 6M / Detector: PIXEL / Date: Sep 22, 2012 Details: KIRKPATRICK-BAEZ PAIR OF BI-MORPH MIRRORS PLUS CHANNEL CUT CRYOGENICALLY COOLED MONOCHROMATOR CRYSTAL |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97857 Å / Relative weight: 1 |
| Reflection | Resolution: 1.41→42.25 Å / Num. obs: 76102 / % possible obs: 99.8 % / Observed criterion σ(I): 2 / Redundancy: 7.2 % / Rmerge(I) obs: 0.07 / Net I/σ(I): 19.2 |
| Reflection shell | Resolution: 1.41→1.47 Å / Redundancy: 7 % / Rmerge(I) obs: 0.65 / Mean I/σ(I) obs: 3.1 / % possible all: 98.8 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 3DCQ Resolution: 1.41→42.24 Å / Cor.coef. Fo:Fc: 0.979 / Cor.coef. Fo:Fc free: 0.971 / SU B: 1.632 / SU ML: 0.029 / Cross valid method: THROUGHOUT / ESU R: 0.055 / ESU R Free: 0.049 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. U VALUES REFINED INDIVIDUALLY. DISORDERED SIDE CHAIN WERE MODELED STEREOCHEMICALLY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 13.07 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.41→42.24 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|